
Гормоны яичка. Патологические состояния детей с недостаточностью гормона яичек
Помимо нарушений дифференцировки клеток Лейдига, в яичках плодов выявлено пять ферментативных дефектов в процессе синтеза тестостерона. Эти дефекты приводят к недостаточной маскулинизации плодов с кариотипом 46,XY. Поскольку и в норме уровень тестостерона до периода полового развития крайне низок, для оценки способности яичек синтезировать этот гормон проводит стимуляционную пробу с ХГЧ.
-
Аплазия клеток Лейдига.
Фенотип больных с аплазией или гипоплазией клеток Лейдига, как правило, женский, хотя возможны и некоторые признаки вирилизации. Обнаруживаются яички, их придатки и семявыносящий проток. Матка и маточные трубы отсутствуют. Большинство вторичных половых признаков в пубертатном возрасте не развиваются, но оволосение лобка может быть нормальным. Уровень тестостерона в плазме низкий и не возрастает при введении ХГЧ, содержание ЛГ повышено. Клетки Лейдига в яичках либо отсутствуют полностью, либо их количество резко снижено. Дефект может локализоваться на уровне рецепторов ЛГ. Стимуляционная проба с ХГЧ позволяет отличить это состояние от синдромов резистентности к андрогенам. Наследование аутосомно-рецессивное, сцепленное с мужским полом. Рецептор ЛГ принадлежит к суперсемейству рецепторов, сопряженных с G-белком, которые содержат семь трансмембранных доменов. У мужчин с гипогонадизмом (предположительно, вследствие гипоплазии или аплазии клеток Лейдига) выявлен ряд инактивирующих мутаций гена рецептора ЛГ.
У одного мужчины с гипогонадизмом, обусловленным мутацией гена Р-субъединицы ФСГ, обнаружен высокий уровень ЛГ и низкий уровень ФСГ в плазме.
-
Липоидная гиперплазия надпочечников.
Эта наиболее тяжелая форма ВГКН получила свое название из-за накопления холестерина и его эфиров в клетках увеличенных надпочечников. Раньше считали, что это результат недостаточности 20,22-десмолазы — фермента, отщепляющего боковую цепь холестерина с превращением его в прегненолон. Теперь же установлено, что липоидная гиперплазия надпочечников — следствие замедленного поступления холестерина во внутреннюю мембрану митохондрий, где функционирует 20,22-десмолаза. Этот процесс зависит от StAR, и у детей с липоидной гиперплазией надпочечников обнаружены мутации его гена.
Уровень всех надпочечниковых стероидов в сыворотке крови либо резко снижен, либо вообще не поддается определению, тогда как содержание АКТГ и активность ренина плазмы повышены. Фенотип женский, производные мюллеровых протоков у мальчиков отсутствуют, поскольку яички вырабатывают фактор регрессии этих протоков, но не продуцируют стероиды. Заболевание проявляется у новорожденных острым гипоадреналовым кризом и синдромом потери соли. Большинство больных имеют кариотип 46,XY. В немногих случаях в период полового развития отмечена продукция эстрогенов. Возможность StAR-независимого стероидогенеза иллюстрируется следующим примером. Из двух 4-месячных близнецов с кариотипом 46,XX и липоидной гиперплазией надпочечников один умер в 15-месячном возрасте от сердечных осложнений коарктации аорты.
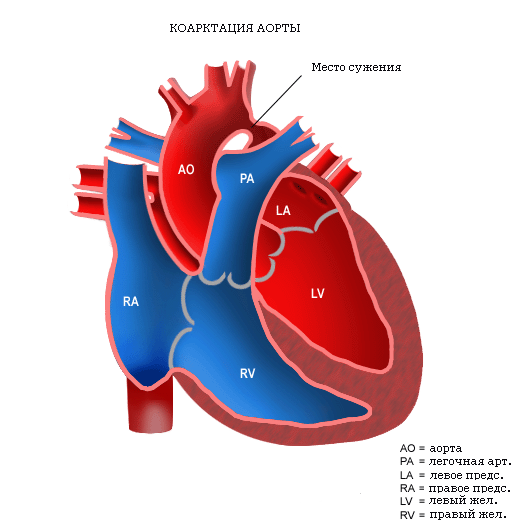
В его надпочечниках обнаруживались типичные отложения липидов. У второго близнеца в 11,5 года началась феминизация; первая менструация отмечена в 13,8 года. При повторном обследовании в 15-летнем возрасте в гене StAR была найдена гомозиготная инактивирующая мутация со сдвигом рамки считывания. Это, а также тот факт, что ребенок до 4-месячного возраста жил без заместительной терапии (и у него определялся альдостерон в сыворотке крови), подтверждает возможность StAR-независимого стероидогенеза, пока переполнение клеток коры надпочечников липидами не нарушит их способности синтезировать стероиды. При частичной недостаточности 20,22-десмолазы у мальчиков наблюдались некоторые признаки вирилизации и позднее начало синдрома потери соли. Полная недостаточность этого фермента, по-видимому, несовместима с жизнью, поскольку только он превращает холестерин в прегненолон, из которого затем образуется прогестерон, необходимый для поддержания беременности. У 4-летнего ребенка с кариотипом 46,XY, половой трансформацией и поздней формой липоидной гиперплазии надпочечников выявлена гетерозиготная мутация гена 20,22-десмолазы. На 6-7-й неделе беременности, когда материнское желтое тело перестает вырабатывать прогестерон, он продуцируется плацентой, в которой StAR не экспрессируется. Таким образом, стероидогенез может поддерживаться ферментной системой P450scc независимо от StAR.

Недостаточность ЗР-гидроксистероиддегидрогеназы. Мальчики с этой формой ВГКН рождаются с той или иной степенью гипоспадии, раздвоенной или нормальной мошонкой, крипторхизмом или без него и только в редких случаях имеют женский фенотип. Признаки синдрома потери соли обычно появляются вскоре после рождения. Наблюдались и случаи частичной недостаточность фермента у мальчиков с преждевременным пубархе к поздней неклассической формой ВГКН. У больных выявлены точечные мутации гена ЗР-гидриксистероиддегидрогеназы типа II, приводящие к нарушению стероидогенеза, которое в надпочечниках и гонадах может быть выражено в разной степени. Нормальное половое развитие у некоторых мальчиков объясняется, вероятно, нормальным функционированием ЗР-гидроксистероиддегидрогеназы типа I в периферических тканях. Больные нередко страдают бесплодием. Описано более 30 разных мутаций гена ЗР-гидроксистероиддегидрогеназы типа II. Тяжесть синдрома потери соли не зависит от степени фенотипических аномалий.
-
Недостаточность 17-гидроксилазы/17,20-лиазы.
Один и тот же фермент, Р450с17, кодируемый геном, расположенным на хромосоме 10q21.3, обладает и 17-гидроксилазной и 17,20-лиазной активностью. Обнаружено много различных дефектов этою фермента. Генетические мальчики обычно рождаются с женским фенотипом и, реже, с той или иной степенью недостаточной вирилизации (от сращения губно-мошоночных складок до промежностной гипоспадии и крипторхизма). Независимо от генетического пола вторичные половые признаки не развиваются.
При классической форме заболевания снижен синтез кортизола в надпочечниках и половых стероидов в надпочечниках и половых железах. Значительно повышенный уровень дезоксикортикостерона в плазме приводит к артериальной гипертонии и гипокалиемии, характерных для этой формы мужского псевдогермафродитизма. Сниженный уровень кортизола компенсируется усиленной секрецией АКТГ и кортикостерона. Дезоксикортикостерин, обладающий минералокортикоидной активностью, подавляет активность ренин-ангиотензиновой системы. Вирилизации в подростковом возрасте не происходит: уровень тестостерона в плазме низкий, а содержание гонадотропных гормонов повышено. Поскольку продукция фактора регрессии мюллеровых протоков во внутриутробном периоде не нарушена, какие-либо остатки этих протоков отсутствуют. При женском фенотипе и генотипе XY показана гонадэктомия и заместительная терапия гидрокортизоном и эстрогенами.
Дефект наследуется аутосомно-рецессивно. При женском генотипе (XX) заболевание обычно диагностируют лишь в пубертатном возрасте, когда не развиваются вторичные половые признаки и проявляется артериальная гипертония и гипокалиемия. Это состояние следует подозревать во всех случаях первичной аменореи и повышения АД у больных с набором хромосом 46,XX или 46,XY.
-
Недостаточность 17-кетостероидредуктазы.
Этот фермент, называемый также 17Р-гидроксистероиддегидрогеназой, катализирует последний этап биосинтеза тестостерона — превращение андростендиона в тестостерон. Он так же катализирует превращение ДЭА в андростендиол и эстрона в эстрадиол. Недостаточность 17-кетостероидредуктазы в ткани яичек плода обусловливает полностью (или почти полностью) женский фенотип при кариотипе 46,XY. Производные мюллеровых протоков отсутствуют, но имеется короткое влагалище. Заболевание диагностируют по соотношению уровней тестостерона и андростендиона в плазме: диагностика до полового развития требует предварительной стимуляционной пробы с ХГЧ.
Дефект наследуется как аутосомно-рецессивный признак. Известно не менее четырех типов 17-кетостероидредуктазы, причем они кодируются различными генами, расположенными на разных хромосомах. Среди арабского населения сектора Газа в Израиле, где распространены близкородственные браки, особенно часто встречается недостаточность фермента типа III. Ген этого фермента локализован на хромосоме 9q22 и экспрессируется только в яичках, где происходит превращение андростендиона в тестостерон. Диагноз в большинстве случаев устанавливается в пубертатном возрасте на основании отсутствия менструаций и появления признаков вирилизации. Содержание тестостерона в плазме может быть нормальным, что объясняется превращением андростендиона в тестостерон в периферических тканях. В это время многие больные осознают себя мужчинами.
Фермент типа I кодируется геном, расположенным на хромосоме 17q21. Он превращает эстрон в эстрадиол и присутствует в плаценте, яичниках, яичках, печени, предстательной железе, жировой ткани и эндометрии. Фермент типа II, ген которого расположен на хромосоме 16q24, катализирует реакции, идущие в противоположном направлении (превращение тестостерона в андростендион и эстрадиола в эстрон) по сравнению с типами I и III. Фермент типа IV действует подобно ферменту типа II. Поздняя форма недостаточности 17-кетостероидредуктазы проявляется гинекомастией у молодых мужчин.
-
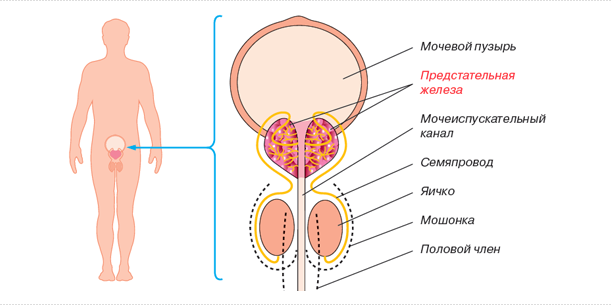
Синдром персистенции мюллеровых протоков.
Это состояние характеризуется сохранением производных мюллеровых протоков при наличии всех остальных признаков вирилизации плода. Описаны случаи такой патологии у родных братьев и однояйцовых близнецов. У 80% больных наблюдается крипторхизм, а обнаружение маточных труб и матки при операции по этому поводу (или по поводу паховой грыжи) позволяет поставить диагноз. Мюллеровы протоки могут быть развиты в разной степени и неодинаково с обеих сторон. Яичники у большинства больных функционируют нормально, но известны и случаи дегенерации этих желез. У некоторых больных после полового развития в яичках развиваются опухоли. При исследовании 38 семей у членов 16 семей были обнаружены дефекты гена, кодирующего фактор регрессии мюллеровых протоков и расположенного на коротком плече хромосомы 19. Уровень этого фактора в плазме был низким. В других 16 семьях с высоким его уровнем выявлен дефект гена, кодирующего рецептор фактора регрессии мюллеровых протоков типа II. У членов 10 из 16 таких семей имелась одинаковая делеция 27 пар оснований в экзоне 10, по крайней мере, в одном из аллелей гена.
Лечение сводится к, по возможности, полному удалению мюллеровых структур без травмирования яичек, их придатков и семявыносящих протоков.
Нарушения действия андрогенов
При этой группе заболеваний синтез тестостерона у плода нормален, а недостаточная вирилизация обусловлена врожденными дефектами действия андрогенов.
-
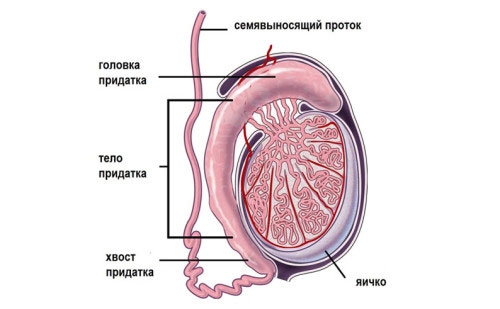
Недостаточность 5а-редуктазы.
Недостаточное образование дигидротестостерона во внутриутробном периоде приводит к резкому нарушению строения наружных половых органов у мужского плода. Биосинтез и периферические эффекты тестостерона при этом сохраняются.
Больные рождаются с очень маленьким половым членом, раздвоенной мошонкой, мочеполовым синусом с промежностной гипоспадией и слепо заканчивающимся влагалищем. Яички имеют нормальное строение и находятся в паховых каналах или губно-мошоночных складках. Производные мюллеровых протоков отсутствуют. Вольфовы структуры (семявыносящие протоки, придатки яичек и семенные пузырьки) сохранены. Большинство больных принимали за девочек. Но в пубертатном периоде у них наблюдается вирилизация: половой член увеличивается, яички опускаются и тоже увеличиваются в размерах, в них происходит сперматогенез. Гинекомастия отсутствует. Волосы на лице растут плохо, предстательная железа маленькая, залысины на висках не образуются. Вирилизация вольфовых протоков обусловлена действием самого тестостерона, но трансформация мочеполового синуса и наружных половых органов зависит от действия дигидротестостерона в критическом периоде их формирования у мужского плода. Рост волос на лице и увеличение предстательной железы, по-видимому, также зависят от дегидротестостерона.
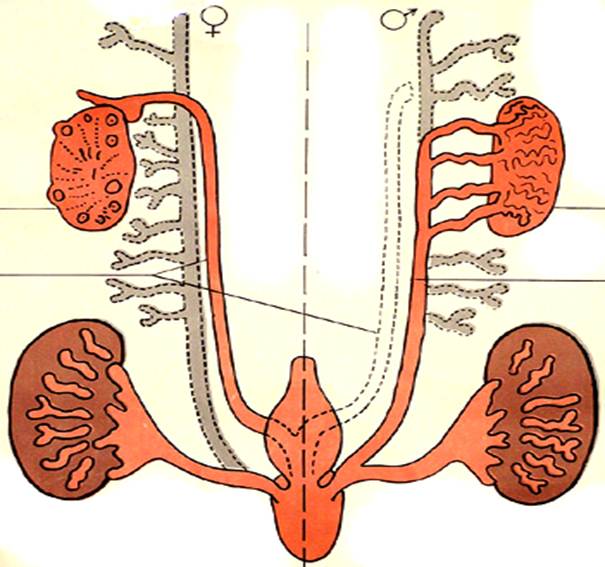
Окончательный рост больных соответствует росту отца и братьев. Однако среди больных существуют значительные фенотипические различия, и в зависимости от фенотипа различают пять типов недостаточности 5а-редуктазы:
-
полностью женский фенотип (тип 5);
-
частично женский (тип 4);
-
промежуточный (тип 3);
-
преимущественно мужской с микропенией (тип 2);
-
полностью мужской (тип 1).
В разных странах у больных обнаружено несколько различных дефектов гена 5а-редуктазы типа 2. Этот ген локализован на коротком плече хромосомы 2. В Доминиканской Республике, Турции, Папуа-Новой Гвинее, Бразилии, Мексике и на Среднем Востоке обнаружена семейная предрасположенность к заболеванию. Фенотип больных не зависит от тяжести генетических дефектов.
Недостаточность фермента наследуется как аутосомно-рецессивный признак, ограниченный мужским полом. Женщины-гомозиготны сохраняют нормальный фенотип и способность к деторождению. Таким образом, дегидротестостерон у женщин не играет роли ни при половой дифференцировке, ни в функции яичников в дальнейшей жизни. Заболевание необходимо диагностировать как можно раньше, причем недостаточность 5а-редуктазы следует отличать от синдрома резистентности к андрогенам. Биохимический диагноз устанавливают на основании нормального содержания тестостерона в крови, нормального или сниженного уровня дегидротестостерона, резко увеличенного отношения тестостерон/дегидротестостерон, особенно после введения ХГЧ (> 17), а также увеличенного отношения этиохоланолон/андростерон и 5р/5а-метаболитов в моче. У детей с резистентностью к андрогенам сохраняется реакция 5а-посстановления в печени и поэтому (в отличие от больных с недостаточностью 5а-редуктазы) отношение тетрагидрокортизол/5а-тетрагидрокортизол остается нормальным.
В большинстве (но не во всех!) случаях дети, воспитываемые как девочки, в период полового развития начинают ощущать себя мужчинами. По-видимому, воздействие тестостерона во внутриутробном, неонатальном и пубертатном периодах играет важную роль в половой самоидентификации мужчин. Однако многое еще предстоит выяснить относительно роли андрогенов, равно как и культурных, социальных, психологических, генетических и других биологических факторов, в половой самоидентификации и половом поведении. Детей с недостаточностью 5а-редуктазы по возможности следует воспитывать как мальчиков. Назначение дегидротестостерона в грудном возрасте приводит к увеличению размеров полового члена.
У фенотипических девочек с неудаленными до пубертатного возраста яичками молочные железы развиваются нормально. Эстрадиол у них синтезируется под действием ароматазы. Определенную роль в феминизации играет и отсутствие андрогенных эффектов.
При частичной резистентности к андрогенам очень трудно выбрать способы психотерапии и хирургических вмешательств. Они зависят в основном от фенотипа больных. Резистентность к андрогенам сопровождается остеопенией.
Как показывают результаты молекулярных анализов, фенотип больных может отчасти зависеть от соматического мозаицизма гена рецептора андрогенов. Так, у больной с кариотипом 46,XY и стоп-кодоном в экзоне 1 этого гена наблюдались признаки вирилизации (оволосение лобка и увеличение клитора), а при тщательном исследовании были обнаружены аллели дикого типа. Мозаицизм сдвигает фенотип в сторону большей вирилизации, чем можно было бы ожидать на основании присутствия в генотипе только мутантного аллеля.
Генетическое консультирование во всех этих случаях сталкивается с большими трудностями. Помимо того, что фенотип не коррелирует с генотипом, в таких семьях часто (27 %) обнаруживаются новые мутации гена рецептора андрогенов.
О тяжести рецепторного дефекта можно судить по степени снижения уровня глобулина, связывающего половые гормоны, после экзогенного введения андрогенов (станозолола). Описаны случаи успешного использования андрогенов при мутациях, приводящих к дефектам ДНК-связывающего и лиганд-связывающего домена рецептора андрогенов.
Мутации гена андрогенного рецептора выявлены также у больных со спинальной амиотрофией и бульбарным параличом, у которых в возрасте 20-50 лет обычно отмечаются атрофия яичек, бесплодие, гинекомастия, а также повышение уровней ЛГ, ФСГ и эстрадиола в сыворотке крови. Мутации этого гена обнаружены и при раке предстательной железы.
Неопределенные причины
Существуют и другие причины недостаточной вирилизации плодов с мужским набором хромосом (XY), демонстрирующие большое разнообразие строения наружных и внутренних половых органов и степени развития мюллеровых структур. Яички могут быть нормальными или рудиментарными, иногда присутствует только одно из них. Установить причину псевдогермафродитизма у многих детей не удается даже с помощью новейших методик. Формирование наружных половых органов промежуточного типа связано с большим количеством хромосомных аберраций, возможность которых необходимо учитывать при дифференциальной диагностике. Наиболее часто встречается синдром 45,X/46,XY. Для выявления мозаицизма может потребоваться исследование нескольких тканей. Различные варианты промежуточного строения наружных половых органов, особенно у мальчиков, могут иметь место и при других сложных генетических синдромах, многие из которых являются результатом мутаций какого-либо одного гена. Такие случаи диагностируют по наличию сопутствующих нарушений развития.
При аутосомно-рецессивном синдроме Смита-Лемли-Опица наблюдаются пре- и постнатальная задержка роста, микроцефалия, птоз, короткий нос с открытыми вперед ноздрями, широкие альвеолярные отростки, синдактилия Н-Ш пальца стопы и тяжелая умственная отсталость. Генетические мальчики обычно рождаются с наружными половыми органами промежуточного типа, а иногда с полной половой трансформацией. Производные мюллеровых протоков, как правило, отсутствуют. У больных с набором хромосом 46,XX наружные половые органы имеют нормальное женское строение. Различают два типа этого синдрома:
-
описанную ранее классическую форму (тип I);
-
акродисгенитальный синдром, который обычно приводит к смерти на первом году жизни и проявляется постаксиальной полидактилией кистей и стоп и резким нарушением строения наружных половых органов (тип II).
Синдрому типа I сопутствует стеноз привратника, а типа II — болезнь Гиршспрунга. При синдроме Смита-Лемли-Опица типа II отмечались расщелина нёба, аномалии скелета, а в 1 случае имелась липома гипофиза.
Для обоих типов синдрома характерен низкий уровень холестерина с повышением содержания его предшественника 7-дегидрохолестерина в плазме.
Мужской псевдогермафродитизм наблюдался также у особей с талассемией и умственной отсталостью.








- Аллергия
- Ангиология
- Болезни глаз
- Венерология
- Гастроэнтерология
- Гинекология
- Дерматология
- Здоровое питание
- Инфекционные болезни
- Кардиология
- Косметология
- Лекарства
- Лекарственные растения
- ЛОР-заболевания
- Мужское здоровье
- Неврология
- Неотложная помощь
- Новости
- Онкология
- Ортопедия
- Паразитология
- Педиатрия
- Пульмонология
- Расшифровка анализов
- Симптомы
- Системные заболевания
- Стоматология
- Травматология
- Урология
- Хирургия
- Эндокринология
- Нужно знать
- Еда
- Профессиональные заболевания
Комментарии