
Врожденные пороки сердца: причины. Роль генетической предрасположенности
Врожденные пороки сердца являются наиболее распространенными пороками развития. Благодаря головокружительным успехам современной кардиохирургии все большее количество пациентов с ВПС доживают до зрелого возраста и сами становятся родителями. В связи с этим особое значение приобретает выяснение степени наследственной обусловленности порока в каждой конкретной семье с последующим расчетом генетического риска.
Как и другие врожденные пороки развития, пороки сердца по этиологическому признаку можно разделить на следующие группы:
-
ВПС, обусловленные наличием хромосомной аномалии;
-
ВПС как часть моногенного синдрома;
-
изолированные пороки сердца с моногенным типом наследования;
-
ВПС, связанные с воздействиями среды;
-
ВПС мультифакториальной природы.
Заподозрить наследственный характер порока сердца может врач любой специальности, однако решение вопроса о степени наследственной обусловленности порока, т.е. отнесение данного конкретного случая к одной из вышеперечисленных групп, должен осуществлять врач-генетик после тщательного всестороннего обследования пациента и членов семьи с применением синдромологического анализа, генеалогического и лабораторных методов исследования.
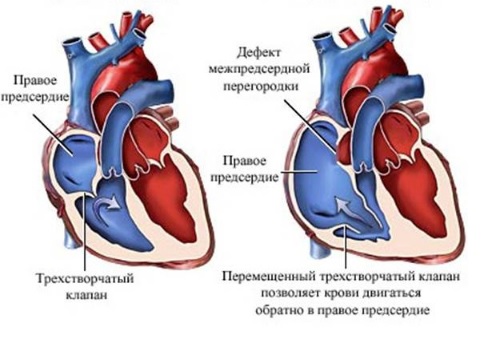
Основным методом, позволяющим заподозрить наследственный характер патологии в данной семье, является генеалогический метод, т.е. метод родословных. Его применение является обязательным для врача любой специальности, который консультирует пациентов с врожденными пороками развития, в том числе и с ВПС.
Суть генеалогического метода заключается в выявлении родственных связей и прослеживании признака или болезней среди близких и дальних, прямых и непрямых родственников. Технически он складывается из двух этапов: составления родословной и генеалогического анализа.
Сбор сведений о семье начинают с консультируемого или пробанда. Дети одной родительской пары называются сибсами. Для графического изображения родословной используют стандартные символы.
Первой задачей при анализе родословной является установление наследственного характера признака. О наследственной природе признака можно думать, если признак встречается в родословной несколько раз и исключается воздействие сходных внешних факторов во время всех беременностей.
Следует помнить, что наличие двух или более случаев ВПС в родословной не является прямым доказательством исключительно наследственной обусловленности порока, а присутствие порока сердца только у пробанда не исключает его наследственной природы.
После составления графической схемы родословной устанавливается тип наследования. Как правило, это делает врач-генетик в медико-генетической консультации или центре с последующим расчетом генетического риска в каждом отдельном случае. Ориентировочно можно учитывать следующие характеристики различных типов наследования.
Наследственно обусловленные ВПС
Наследственно обусловленные ВПС представляют собой гетерогенную группу и включают:
1. Моногенные ВПС, которые наследуются согласно законам Менделя. Они представлены:
-
изолированными пороками сердца при отсутствии врожденных пороков или аномалий развития других органов и систем. Так, описаны случаи аутосомно-доминантного наследования мутации специфичного гена Мкх2-5 на хромосоме 5д35, которая является ответственной за развитие вторичного ДМПП и нарушение AV-проводимости;
-
синдромами множественных врожденных пороков развития, в которых ВПС является одним из составляющих синдром признаков. Моногенное наследование касается всего синдрома как единого целого, однако проявление может быть у разных членов семьи неполным и колебаться от одного-единственного признака до разной комбинации составляющих этот синдром врожденных пороков развития.
2. Хромосомные синдромы, причиной возникновения которых служит изменение количества и/или структуры хромосом.
Если ВПС являются частью генного или хромосомного синдрома, у подавляющего большинства пациентов имеются и другие пороки развития, нарушения физического и психического развития различной степени тяжести, нарушения метаболизма и/или другая сопутствующая патологию. По данным проведенных исследований, до 66% врожденных пороков развития у плодов и мертворожденных детей носит характер множественных.
В большинстве случаев моногенные синдромы с ВПС относятся к группе синдромов с МВПР, и для их диагностики применяется клинико-генеалогический метод с синдромологическим анализом. Также применяются молекулярно-генетические методы – прямые, в случае известной нуклеотидной последовательности патологического гена, и непрямые, когда нуклеотидная последовательность еще не известна и вместе с тем имеется информация об относительном положении гена на генетической карте. Биохимические методы применяются редко, однако в некоторых случаях они являются ведущими. Так, например, биохимический метод лежит в основе подтверждающей диагностики синдрома Смита-Лемли-Опитца, в спектре клинического проявления которого присутствует ВПС. Для дифференциальной диагностики применяются и цитогенетические методы, так как генные синдромы с МВПР и хромосомные синдромы в ряде случаев имеют сходную клиническую картину.
Медико-генетическое консультирование при моногенных синдромах основывается на установлении типа наследования данного заболевания с последующим расчетом генетического риска для сибсов и потомков с определением наиболее эффективного способа профилактики.
Подавляющее большинство ассоциаций являются спорадическими случаями, т.е. риск для сибсов не превышает общепопуляционный.
Хромосомные синдромы
Хромосомные синдромы – группа врожденных патологических состояний, в основе которых лежат геномные или хромосомные мутации.
Геномные мутации, или количественные изменения хромосомного набора, могут быть двух типов:
-
полиплоидии – увеличение числа хромосом, кратное гаплоидному набору хромосом ;
-
анеуплоидии – увеличение или уменьшение числа хромосом в наборе, некратное гаплоидному.
К хромосомным мутациям относятся структурные перестройки одной или двух и более хромосом, которые могут затрагивать всю хромосому или ее часть. С точки зрения цитогенетики, структурные перестройки хромосом классифицируются по принципу линейной последовательности расположения генов:
-
делеции;
-
инверсии;
-
инсерции;
-
транслокации;
-
кольцевые хромосомы;
-
изохромосомы.
По данным Schinzel, ВПС относятся к одним из частых пороков развития, сопровождающих хромосомные аномалии. Согласно базе данных CARIS, у 12% детей с ВПС имеются те или иные хромосомные аномалии. В 70% случаев это трисомии по хромосомам 21, 18 или 13.
Хромосомный синдром у пациента с ВПС можно заподозрить по следующим признакам:
-
врожденные пороки других органов и систем;
-
задержка психомоторного и физического развития;
-
задержка речевого развития, нарушение процесса познания, необычное поведение, нарушение внимания с гиперактивностью и другие нарушения психологического статуса;
-
наличие у пациента необычных черт лица, что делает его непохожим на родителей и сибсов, особенно при наличии отставании в развитии;
-
малая масса пациента при рождении, преждевременные роды или признаки незрелости при родах в срок;
-
наличие хромосомных аномалий у родителей или сибсов;
-
дополнительным критерием может быть наличие в анамнезе родителей длительного бесплодия, самопроизвольных абортов на ранних сроках, мертворождений.
Выявление вышеописанных признаков требует направления пациента в медико-генетический центр и идентификации хромосомных аномалий цитогенетическим методом. Основой данного метода является исследование кариотипа человека.
Кариотип – это совокупность морфологических особенностей полного хромосомного набора, типичная для клеток представителя данного биологического вида. Специфичность кариотипа определяется общим числом хромосом, их размером и формой, а количественная и структурная стабильность хромосом является важнейшим условием формирования фенотипически нормального организма в ходе индивидуального развития. У человека диплоидный набор хромосом в соматических клетках составляет 46. Целью хромосомного анализа в клинической цитогенетике является оценка кариотипа с выявлением возможных количественных или структурных аномалий хромосом путем анализа метафазных или прометафазных препаратов хромосом и правильная запись кариотипа в соответствии с унифицированной системой записи и символизации.
-
Синдром Дауна.
Синдром Дауна является наиболее частым хромосомным синдромом у пациентов с ВПС. Синдром хорошо описан в многочисленных руководствах. Пороки сердца присутствуют более чем у 50% детей с синдромом Дауна и являются основной причиной гибели этих пациентов. Наиболее типичными являются АВСД, ДМЖП, ДМПП, ОАП, аберрантная подключичная артерия.
-
Синдром Патау.
Причиной является трисомия по хромосоме 13. Синдром сопровождается МВПР развития и ранней гибелью пациентов. Средняя продолжительность жизни составляет 2,5 дня, менее чем 3% пациентов доживают до 6 мес.
Наиболее характерной комбинацией врожденных пороков развития является сочетание постаксиальной полидактилии, несращения губы и неба, микрофтальмии, колобомы глаз и голопрозэнцефалии. Кроме того, специфическими являются врожденные пороки сердца и мочеполовой системы.
-
Синдром Эдвардса.
Врожденные пороки сердца являются характерным признаком и третьей, наиболее частой, трисомии – трисомии по хромосоме 18 . Как и синдром Патау, синдром Эдвардса относится к полулетальным синдромам. Более 90% пациентов умирают, не доживая до полугода.
Основными признаками синдрома Эдвардса являются: резкая пренатальная гипоплазия, долихоцефалия, открытый метопионический шов, нависающий затылок, низко расположенные, ротированные кзади, диспластические ушные раковины, маленький рот, микрогнатия, короткая грудина, флексорное положение конечностей, перекрывание третьего пальца руки вторым и четвертого пальца пятым, гипоплазия ногтей, стопа-качалка, синдактилия П-Ш на стопах. Из пороков внутренних органов наиболее часто встречаются пороки развития головного мозга, пороки сердца, почек и желудочно-кишечного тракта. Пороки сердца выявляются более чем у 90% детей с синдромом Эдвардса. Наиболее характерными являются ДМЖП, ДМПП, ДВПЖ. Также часто встречаются пупочные грыжи, атрезии пищевода с трахеоэзофагальным свищом, общая брыжейка, единственная подковообразная почка, поликистоз почек, гидронефроз. Почти у 10% пациентов наблюдается аплазия лучевой кости и/или большого пальца кисти. Основными пороками развития, позволяющими заподозрить наличие синдрома Эдвардса, являются трахеоэзофагальный свищ, аплазия лучевой кости, пороки сердца, незавершенный поворот кишечника. Трисомии как по хромосоме 13, так и по хромосоме 18 относятся к полулетальным синдромам с множественными пороками развития. При подтвержденном цитогенетическом диагнозе в случае согласия родителей в большинстве клиник отказываются от проведения хирургической коррекции пороков развития, обеспечивая симптоматическое лечение.
-
Синдром Шерешевского-Тернера.
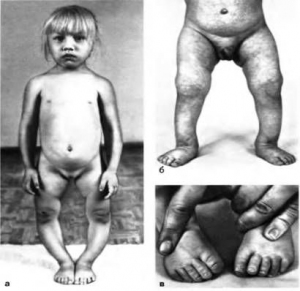
Пороки сердца встречаются и при нарушении количества половых хромосом. Врожденные пороки сердца присутствуют более чем у 1/3 пациенток с синдромом Шерешевского-Тернера. Наиболее типичными являются коарктация аорты, двустворчатый аортальный клапан, атрезия клапана аорты. Для периода новорожденности характерны следующие признаки: малая масса при рождении, лимфатический отек тыльной поверхности кистей и стоп, гипоплазия ногтей, крыловидные складки на шее, широкая грудная клетка, иногда вдавленная, с широко расставленными гипоплазированными сосками. С возрастом задержка роста становится все более заметной, однако в ряде случаев врачи связывают ее с наличием у пациента ВПС. В период полового созревания основными диагностическими признаками синдрома Шершевского-Тернера являются: половой инфантилизм, аменорея, низкий рост. В большинстве случаев диагноз ставится именно в пубертатный период. Следует отметить, что деформация грудной клетки, которая часто при наличии порока сердца оценивается в качестве вторичного изменения, также является одним из характерных признаков синдрома. На 2-м десятилетии жизни возможно развитие артериальной гипертензии. Планируя тактику хирургического лечения ВПС, следует учитывать, что при синдроме Шершевского-Тернера в ряде случаев описано развитие рекоарктации аорты через несколько лет после проведенной операции.
-
Синдромом Вольфа-Хиршхорна.
Врожденные пороки сердца имеют место также более чем у 33% пациентов с синдромом Вольфа-Хиршхорна и являются основной причиной гибели таких пациентов. Характерные признаки этого синдрома: лицо в виде греческого шлема с выступающими лобными буграми, выступающее надпереносье, гипертелоризм, гипоплазия надбровных дуг, эпикант, широкий клювовидный нос. Также встречаются пороки наружных половых органов и у 15% больных – несращения губы и/или нёба. Кроме того, у подавляющего большинства таких больных наблюдаются грубая задержка развития и судороги. Врожденные пороки сердца часто комбинированные, включают пороки развития клапанов, персистирование левой верхней полой вены, а также ДМЖП, ДМПП, ОАП, стеноз легочной артерии. Для синдрома Вольфа-Хиршхорна характерны также пороки развития головного мозга и почек. Хотя ВПС в большинстве случаев поддаются коррекции, отдаленный прогноз для жизни часто сомнительный, более трети пациентов умирают в возрасте до 1 года. Тяжелое течение послеоперационного периода объясняется также наличием МВПР и неврологическими нарушениями.
В последние годы в связи с развитием молекулярно-цитогенетических методов диагностики в отдельную группу выделили так называемые микроструктурные аномалии хромосом. К этой группе относятся аномалии строения хромосом, которые не выявляются при рутинном цитогенетическом исследовании вследствие своих малых размеров. Для их обнаружения требуется проведение молекулярно-цитогенетических исследований с использованием специфических ДНК-зондов.
Из этой группы синдромов необходимо выделить синдром микроделеции 22q11.2 и синдром Вильямса.
-
Синдром микроделеции 22q11.2.
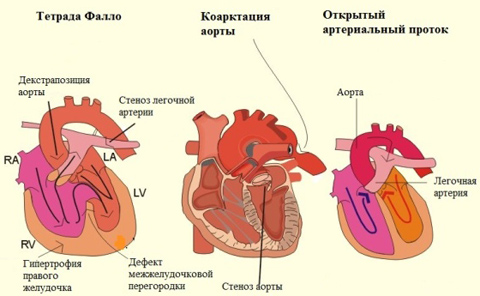
Синдром микроделеции 22q11.2, или CATCH22- синдром, встречается у 1 из 4000 живых новорожденных и является вторым хромосомным синдромом после синдрома Дауна, для которого пороки сердца служат диагностическим признаком. Наиболее характерными являются конотрункальные пороки сердца – прерванная дуга аорты, артериальный ствол, атрезия легочной артерии. Также часто встречаются тетрада Фалло, ДМЖП, аберрантная правая подключичная артерия. Так, почти 50% пациентов с ОАС имеется данная хромосомная аномалия, т.е. выявление у пациента этого порока сердца требует проведения молекулярно-цитогенетического исследования, независимо от наличия или отсутствия других симптомов. Другими характерными признаками являются: малые лицевые аномалии, несращение нёба, гипокальциемия, гипоплазия тимуса с нарушением клеточного иммунитета. Следует отметить, что гипокальциемия присутствует чаще только на 1-м году жизни и носит латентный характер. Однако при различных стрессовых воздействиях уровень кальция может снижаться и сопровождаться появлением судорожного синдрома. Данную особенность необходимо учитывать при подготовке больного к оперативному вмешательству. В редких случаях развитие гипокальциемии возможно и у взрослых пациентов в состоянии полного здоровья.
-
Синдром Вильямса.
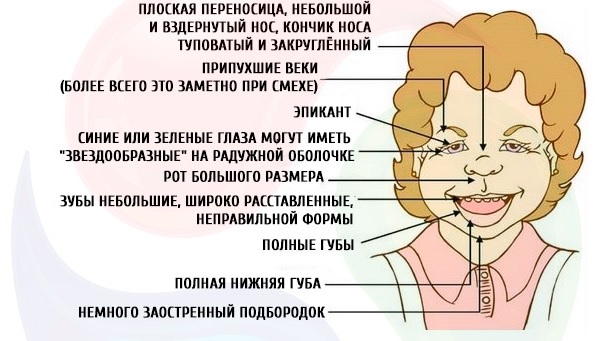
Причиной развития синдрома Вильямса является микроделеция по длинному плечу хромосомы 7-7ц11.23. При проведении молекулярно-цитогенетического исследования она обнаруживается более чем у 99% пациентов. Синдром Вильямса характеризуется лицевым дизморфизмом, наличием ВПС, нарушением умственного развития, идиопатической гиперкальциемией в течение первого года жизни. Наиболее характерными пороками сердца являются надклапанный стеноз аорты и периферические стенозы легочной артерии. Причем надклапанный стеноз аорты манифестирует чаще во 2-м полугодии. Следует отметить, что основные клинические признаки синдрома Вильямса, а именно: лицевой дизморфизм и задержка развития – могут отсутствовать в неонатальном периоде, и ВПС может быть единственным симптомом. В связи с этим рекомендуется всем пациентам с надклапанным стенозом аорты, особенно в раннем возрасте, проводить специальное молекулярно-генетическое исследование. Особого внимания и своевременной диагностики данный синдром требует в связи с тем, что у этих пациентов повышен риск развития синдрома внезапной смерти при проведении общего наркоза.
Для хромосомных синдромов характерно отсутствие менделевского типа наследования. Преимущественно патология возникает спорадически "de novo", являясь следствием мутации в гамете одного из родителей. В таких случаях риск для сибсов пациента, т.е. риск повторного рождения ребенка с хромосомным синдромом, в данной семье невелик и не превышает общепопуляционного. Однако часть хромосомных синдромов может наследоваться, если один из родителей является носителем хромосомной перестройки. В части случаев носители сбалансированного кариотипа фенотипически нормальны, однако для потомков существует большой риск иметь несбалансированный кариотип. В других случаях носители имеют врожденные пороки развития или множественные стигмы с нарушением нервно-психического развития, однако фертильность у них сохранена. Риск для потомков родителя-носителя сбалансированного кариотипа значительно варьирует в зависимости от вовлеченной в перестройку хромосомы и локализации точек разрыва.
Воздействие внешней среды на плод
Врожденные пороки сердца, связанные с воздействиями среды, выступают как одно из проявлений эмбриопатии или фетопатии. Таким образом, воздействие тератогенов в критические периоды развития различных анатомических структур сердца и крупных сосудов наиболее опасно, так как несет риск возникновения врожденных пороков.
Наиболее важными в практике являются хронические заболевания у матери и прием женщиной лекарственных средств или других химических тератогенов.
Описано и возникновение ВПС у плода при наличии приобретенных пороков сердца у матери, особенно при активации ревматического процесса.
При фетальном алкогольном синдроме в определенном проценте случаев также встречаются пороки сердца. Наиболее частыми являются ДМЖП, также имеют место ДМПП и тетрада Фалло. Фетальный алкогольный синдром развивается только у 40% женщин, употребляющих алкогольные напитки в I триместре беременности, причем критической дозы алкоголя до настоящего времени не установлено. Наиболее характерными для фетального алкогольного синдрома являются короткие глазные щели, длинный плоский фильтр, тонкая верхняя губа со сглаженной линией Купидона, характерная ладонная складка в виде клюшки для гольфа; кроме того, у детей с фетальным алкогольным синдромом наблюдается грубая задержка развития.
Изолированные случаи ВПС, т.е. пороки сердца, не сопровождающиеся пороками других органов, нарушением психофизического развития и другой патологией, в большинстве случаев имеют мультифакториальную природу. Мультифакториальная природа порока сердца подразумевает, что для реализации фенотипа необходимо присутствие генетической и средовой компоненты одновременно. Генетическая компонента представляет собой комплекс нескольких генов, каждый из которых выполняет определенную функцию, а вместе они контролируют нормальное развитие органа. При возникновении определенного количества изменений в этих генах или получения мутантных генов от родителей генетическая компонента возрастает и может быть значимой в процессе развития ВПС. Средовая компонента – это различные воздействия среды на ранних этапах внутриутробного развития, которые сами по себе не приводят к формированию порока сердца, а реализуются только при соответствующем генотипе, т.е. при наличии генетической компоненты.
Расчет риска при пороках сердца мультифакториальной природы осуществляется по таблицам эмпирического риска, которые представлены в специальных руководствах. Риск выше при более близком родстве с пациентом с наличием ВПС, а также повышается при нескольких случаях пороков сердца в данной родословной.
Профилактика ВПС
К методам профилактики возникновения ВПС, как и других врожденных пороков развития, можно отнести назначение фолиевой кислоты женщине на протяжении 3 мес. до наступления беременности и в течение всего I триместра. Доза фолиевой кислоты составляет 400 мкг/сут, при отягощенном анамнезе суточная доза повышается в 10 раз и составляет 4 мг/сут.
Выявление ВПС у плода возможно с помощью ультразвуковой пренатальной диагностики, начиная уже со II триместра. Основной целью ультразвукового скрининга II триместра и является диагностика врожденных пороков развития, в том числе ВПС у плода.
Выявление определенных типов врожденных пороков развития может предполагать наличие у плода наследственного синдрома. Так, во всех случаях пренатальной диагностики АВСД необходимо осуществление инвазивного вмешательства – амниоцентеза либо кордоцентеза – в целях проведения цитогенетического исследования материала плода для исключения синдрома Дауна. В случаях пренатального выявления тетрады Фалло у плода рекомендовано проведение молекулярно-цитогенетического исследования для исключения синдрома микроделеции 22q11.2.
В заключение необходимо еще раз отметить, что диагностика наследственной патологии и расчет индивидуального генетического риска в каждом конкретном случае должны проводиться врачом-генетиком в специализированном учреждении – медико-генетическом центре или медикогенетической консультации.
Основные признаки, позволяющие заподозрить наследственную обусловленность ВПС и требующие уточнения этиологии конкретного случая:
-
Присутствие 2 и более случаев ВПС в семье.
-
Сочетание порока сердца с врожденными пороками других органов и систем.
-
Сочетание ВПС с нарушением психомоторного и физического развития.
-
Задержка речевого развития, нарушение процесса познания, необычное поведение, нарушение внимания с гиперактивностью и другие нарушения психологического статуса.
-
Наличие у пациента необычных черт лица, что делает его непохожим на родителей и сибсов, особенно при наличии отставания в развитии.
-
Сочетание ВПС с кожными изменениями, необычным запахом от пациента, изменением цвета мочи или другими специфическими, редко встречающимися симптомами.
-
Наличие хромосомных аномалий у родителей или сибсов или наличие больного моногенным синдромом в семье.
-
Наличие в анамнезе родителей длительного бесплодия, самопроизвольных абортов на ранних сроках, мертворождений.








- Аллергия
- Ангиология
- Болезни глаз
- Венерология
- Гастроэнтерология
- Гинекология
- Дерматология
- Здоровое питание
- Инфекционные болезни
- Кардиология
- Косметология
- Лекарства
- Лекарственные растения
- ЛОР-заболевания
- Мужское здоровье
- Неврология
- Неотложная помощь
- Новости
- Онкология
- Ортопедия
- Паразитология
- Педиатрия
- Пульмонология
- Расшифровка анализов
- Симптомы
- Системные заболевания
- Стоматология
- Травматология
- Урология
- Хирургия
- Эндокринология
- Нужно знать
- Еда
- Профессиональные заболевания
Комментарии