
Иммунодефицитные состояния. Синдромы иммунного дефицита
На первый взгляд, связь между иммунодефицитными расстройствами и новообразованиями лимфоидной системы неотчетлива, поэтому возникает вопрос о том, целесообразно ли их обсуждать в одной статье. Для иммунодефицитных синдромов характерно отсутствие или недостаточность, в то время как новообразование отражает избыток или неконтролируемую пролиферацию. Однако связь между ними является примером того, насколько тонко настроена и сложна иммунная система. Как описывается в настоящей главе, дефициты, особенно в одном из звеньев иммунной системы, нарушают способность оставшихся элементов контролировать собственный рост. По этой причине иммунодефициты являются благодатной почвой для развития новообразований. Это не касается дефектов клеток- предшественников или ранних стволовых клеток, которые приводят к более выраженным повреждениям функций иммунитета. Аутоиммунные феномены являются более выраженными проявлениями потери иммунной регуляции, которая часто сопровождает иммунодефицита. Таким образом, три казалось бы, несвязанных болезненных состояния — иммунодефицит, аутоиммунные заболевания и лимфоидные новообразования — часто сосуществуют в одном организме.

Как известно, иммунный ответ опосредуется Т- и В-лимфоцитами, натуральными киллерами (NK), клетками миелоидного/моноцитарного ростка и комплементом. Взаимодействия между этими клетками, выделяемые ими растворимые медиаторы (антитела и цитокины) и система комплемента находятся под строгим контролем. Расстройства развития и дифференцировки этих клеток, синтеза ими своих продуктов, нарушения взаимодействий между ними могут привести к иммунодефицитным состояниям, клиническая тяжесть которых варьирует от легких до фатальных.
Однако существенная часть дефицитов не проявляется клинически, поскольку затрагивает те отделы, где иммунная система проявляет избыточность, так, в участке цитокиновой сети функции одного компонента могут быть замещены другим.
Хотя врожденные иммунодефицитные заболевания (состояния, с которыми люди рождаются) в основном редки, описания ранних стадий этих «экспериментов природы» пролили свет на функционирование иммунной системы. Эксперименты на животных, в которых имитируются различные типы иммунодефицитов человека, показали, что клетки специфического иммунитета делятся на Т- и В-лимфоциты, т.е. клеточно-опосредованный иммунитет против гуморального.
В настоящее время эти редкие синдромы иммунодефицита и случаи лимфоидных новообразований анализируются на молекулярном уровне. Информация, полученная в ходе исследований этих заболеваний, применяется для их лечения и разработки иммунологических препаратов для аутоиммунных заболеваний и нелимфоидных злокачественных опухолей. Данная глава начинается с описания врожденных и приобретенных синдромов иммунного дефицита, а завершается рассказом о новообразованиях иммунной системы.
Синдромы иммунного дефицита
Иммунодефипитные состояния разделяются на две основные группы:
-
первичные, которые могут быть наследственными или приобретенными, но причиной заболевания является именно иммунный дефицит;
-
вторичные, при которых иммунный дефицит является результатом других болезней или состояний.
Первичные иммунные дефициты можно классифицировать, основываясь на клинических проявлениях, что соотносится с поражением определенного звена иммунной системы:
-
Т-клеток, или клеточно-опосредованного иммунитета;
-
В-клеток, или антитело-опосредованного иммунитета;
-
Т- и В-клеточного иммунитета;
-
неспецифического иммунитета, опосредованного фагоцитирующими клетками и/или NK-клетками;
-
активацией комплемента.
Эта классификация упорядочивает широкий спектр иммунных расстройств.
Аномалии цитокинов и цитокиновых рецепторов — средств, с помощью которых клетки обмениваются информацией и функционируют, не являются отдельной категорией расстройств, а входят в первые четыре группы. Поскольку проявляющийся иммунный ответ часто является результатом взаимодействия клеток нескольких типов, недостаточность, например, продукции антител и функций В-клеток может на самом деле быть обусловлена первичным нарушением Т-клеток или Т- В-клеточного взаимодействия. Классификация, основанная на видимом проявлении дефекта и не нуждающаяся в выявлении его первопричины (которая может быть неизвестна), является удобным основанием для диагностики новых случаев заболевания. Эта классификация также позволяет выявить корреляцию с экспериментальными моделями на животных, с помощью которых может быть идентифицирован основополагающий иммунный дефект.
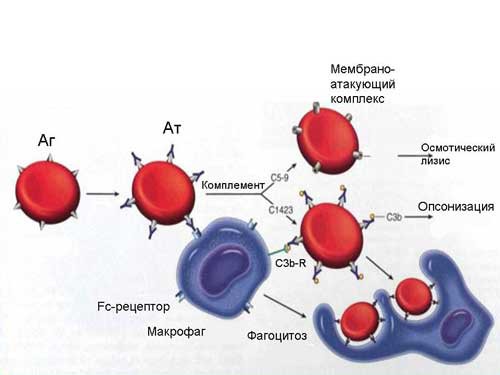
Наличие иммунодефицита можно заподозрить у пациента, страдающего повторными инфекционными заболеваниями. Тип обнаруживаемой инфекции часто может облегчить диагностику причины иммунодефицита. Например, повторяющиеся бактериальные средние отиты (инфекции уха) и бактериальные пневмонии часто встречаются у лиц с недостаточностью В-клеток и антител. Повышенная восприимчивость к грибковым, протозойным и вирусным инфекциям наблюдается при недостаточности Т-клеток и клеточно-опосредованного иммунитета. Системные инфекции, вызванные бактериями с нормальной или сниженной вирулентностью, поверхностные инфекции кожи или инфицирование пиогенными (образующими гной) микроорганизмами предполагают недостаточность фагоцитирующих клеток; повторные инфекции пиогенными организмами связаны с дефицитом комплемента. Особое значение имеет развитие оппортунистических инфекций, т.е. болезней, вызванных микроорганизмами, обычно присутствующими в окружающей среде и непатогенными для иммунокомпетентных лиц. Среди микроорганизмов, с наибольшей частотой участвующих в развитии таких заболеваний, можно назвать Pneumocystis carinii, цитомегаловирус (CMV), токсоплазмы, Mycobacterium avium и различные виды Candida. Чаще всего такие инфекции связаны с недостаточностью клеточно-опосредованного иммунитета.
За исключением недостаточности IgA распространенность синдромов первичного иммунодефицита очень низкая — порядка одного случая на 10000 чел. Примерно 50% всех случаев составляет недостаточность антител, 20% приходится на комбинированную недостаточность иммунитета (и по антителам, и по клеточно-опосредованным механизмам), 18% составляют расстройства фагоцитоза, 10% — это нарушения только клеточно-опосредованного иммунитета и 2 % составляют дефициты комплемента. Чем раньше при развитии плода возникает генетический дефект или блок, тем больше звеньев иммунной системы оказываются пораженными и тем более тяжело протекает болезнь.
Тяжелый комбинированный иммунодефицит
Тяжелый комбинированный иммунодефицит (ТКИД) — гетерогенная группа заболеваний, при которых дефектными являются одновременно и клеточно-опосредованный иммунитет, и продукция антител. Это состояние первоначально называли а-гаммаглобулинемией швейцарского типа. Пациенты с ТКИД гипотетически восприимчивы ко всем типам микробной инфекции (вирусной, бактериальной, грибковой и протозойной), особенно — CMV, Pneumocystis carinii и Candida. Вакцинация ослабленным живым вирусом у таких младенцев может привести к фатальным последствиям.
Заболевание может быть классифицировано по подгруппам при первичном обследовании в соответствии с присутствием в крови определенных субпопуляций лимфоцитов. В одной из подгрупп, обозначаемой Т В+, практически отсутствуют Т-клетки, но присутствует нормальное или повышенное количество нефункционирующих В-клеток. У больных этой подгруппы может не быть и NK-клеток. Во второй подгруппе, Т В, отмечается тяжелая лимфопения в связи с отсутствием как Т, так и В-клеток. Некоторые больные попадают в подгруппу Т+В+, и лишь единичные — в подгруппу Т+В. Предпочтительным методом лечения для всех пациентов с ТКИД является пересадка костного мозга, из которого удалены (деплетированы) Т-клетки, от совместимого по HLA родственного донора.
-
Подгруппа В+.
V-сцепленный ТКИД. Пациенты с ТКИД, сцепленным с хромосомой X, составляют 40-50 % случаев ТКИД; в большинстве случаев проявлением заболевания служит Т В+-лимфопения. Были обнаружены мутации в гене, расположенном на хромосоме X, который кодирует у-цепь, которая является общей в рецепторах для цитокинов IL-2, IL-4, IL-7, IL-9 и 1L-15. Таким образом, при этой мутации нарушается ответ на множество цитокинов.
При описании заболеваний человека очень информативным оказалось исследование на экспериментальных животных, у которых были смоделированы дефекты иммунной системы. У «нокаутных» мышей отсутствовала у-цепь, так же как и у пациентов с ТКИД, и обнаруживался дефект развития как Т-, так и В-клеток. Мыши, у которых отсутствовали IL-7 и IL-7R, также походили на пациентов с ТКИД, что позволяет предположить, что IL-7 необходим для развития Т- и NK-клеток и что его функции не могут быть компенсированы другими цитокинами. У «нокаутных» мышей, не имеющих гена IL-2, обнаруживалась только некоторая дисфункция иммунной системы с нормальным развитием Т- и В-клеток и отсутствием фенотипа ТКИД.
Аутосомно-рецессивный ТКИД. Небольшая подгруппа пациентов, для которых характерна Т В+-лимфопения, демонстрирует скорее аутосомно-рецессивный, чем сцепленный с хромосомой X тип наследования. Фенотип таких пациентов и больных с Х-сцепленным ТКИД идентичен, и клинически эти подгруппы разделить невозможно. Мутации локализуются в гене тирозиновой киназы JAK3, внутриклеточной молекулы, ответственной за передачу сигналов от у-цепь цитокиновых рецепторов. Обычно JAK3 экспрессируется только в гемопоэтических клетках
-
Подгруппа ТВ.
Дефицит аденозиндезаминазы. Аденозиндезаминаза (АДА) — фермент цикла утилизации пуринов, является повсеместно экспрессируемым ферментом «домашнего хозяйства» (т.е. используется при постоянном функционировании клетки). Пациенты, у которых отсутствует этот фермент, составляют примерно 20 % больных с ТКИД; заболевание имеет аутосомно-рецессивный механизм наследования. Недостаточность АДА приводит к накоплению токсических отходов метаболизма, вызывая с течением времени прогрессию симптомов. Такая картина делает раннюю диагностику и назначение лечения в этой группе больных жизненно важными.
Дефицит АДА оказывает самое выраженное влияние на иммунную систему, так как приводит к недостаточности развития как Т, так и В-лимфоцитов. У многих пациентов имеются характерные аномалии скелета. Не совсем понятно, почему у них не развиваются более выраженные мультиорганные проблемы. Исследования такого редкого генетического заболевания показали особенную важность этого «спасательного пути» для развития и дифференцировки лимфоцитов. Также они привели к разработке противолейкозных препаратов, предотвращающих рост злокачественных лимфоцитарных предшественников. Пациенты с дефицитом АДА, у которых не было подходящего родственного донора костного мозга, были первой группой больных, которых лечили с помощью генной терапии, заключавшейся во введении с помощью вируса функционального гена АДА. Однако даже после многих лет совершенствования этот экспериментальный подход все еще сопряжен с трудностями. Альтернативным методом лечения является дотационная ферментотерапия.
Дефицит фосфорилазы пуриновых нуклеозидов. Мутация в гене другого фермента пути утилизации пуринов, фосфорилазы пуриновых нуклеозидов (ФПН), сходным образом приводит к избытку токсических продуктов, особенно сильно повреждающих нервную систему и Т-клетки. Вследствие этого дефекта во всех лимфоидных тканях — тимусе, миндалинах, лимфатических узлах и селезенке, наблюдается выраженный дефицит лимфоцитов. Парадоксальным образом, даже несмотря на заметный иммунодефицит у детей с этим состоянием, у таких пациентов часто встречаются аутоиммунные заболевания.
Недостаточность рекомбиназ. Гены, активирующие рекомбинации (RAG), 1 и 2 кодируют ферменты, участвующие в реаранжировке генов Ig в пре-В-клетках и генов Т-клеточного рецептора в пре-Т-клетках. Оба фермента необходимы для этих реаранжировок, поэтому мутация в любом из них приводит к полному отсутствию Т-клеток, В-клеток и иммуноглобулинов. Созревание останавливается на стадиях пре-Т- и пре-В-клеток. Обычно функция NK-клеток не нарушается.
-
Подгруппа Т+В.
Синдром Оменна. Синдром Оменна является «текучим» ТКИД со сниженной, но частично сохраненной активностью RAG. Природа этого заболевания все еще до конца не изучена. Клинические проявления у пациентов больше всего похожи на тяжелые проявления реакций «трансплантат против хозяина» (РТПХ), чем на симптомы у пациентов с отсутствием активности RAG. Несмотря на тяжелые проявления иммунодефицита, которые препятствуют формированию эффективного иммунного ответа на любой возбудитель, у таких пациентов обнаруживается выраженная дисрегуляция иммунной системы. Лимфоциты периферической крови этих больных имеют фенотип Т+В, а в коже и ЖКТ отмечается массивная инфильтрация эозинофилами и активированными Т-клетками, выделяющими цитокины типа Тн2. Это приводит к гипер-IgЕ-синдрому и нарушению питания в связи с потерей белка. По сравнению с пациентами с другими типами ТКИД степень успешности трансплантации костного мозга у людей с синдромом Оменна невелика. Причиной неудач является отторжение трансплантата. Таким образом, несмотря на то, что пациенты с синдромом Оменна страдают иммунодефицитом, они нуждаются в иммуносупрессивной терапии.
-
Подгруппа Т+В+.
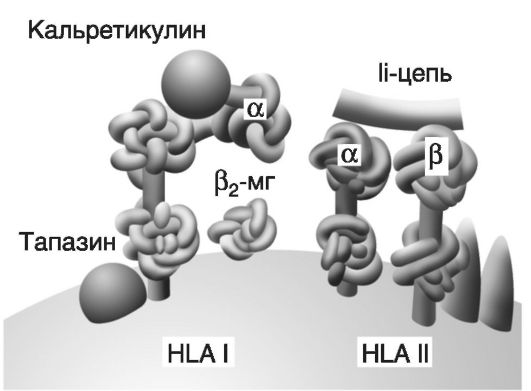
Синдром «голых» лимфоцитов. Синдром «голых» лимфоцитов (СГЛ) развивается в результате нарушения экспрессии молекул HLA (человеческого МНС). Его разделяют на три группы в зависимости от того, молекулы HLA какого класса отсутствуют: I, II или одновременно обоих. Несмотря на это, признаки постоянного иммунодефицита возникают только у пациентов, не экспрессирующих молекулы HLA II класса вне зависимости от того, экспрессируют они молекулы HLA I класса или нет. Количество циркулирующих Т- и В-клеток может быть нормальным; однако при отсутствии молекул HLA II класса белковые антигены не могут быть презентированы СD4+-Т-клеткам. Поэтому не возникает взаимодействия между АПК (В-клетками, макрофагами/моноцитами. дендритными клетками) и CD4+-T- клетками. Вследствие этого не обеспечивается поддержка функционирования В-клеток или образования цитотоксических Т-клеток. Это приводит к клиническим проявлениям комбинированного иммунодефицита. Поскольку экспрессия молекул МНС II класса на эпителиальных клетках тимуса необходима для позитивного отбора С04+-Т-клеток, то таких клеток в тимусе образуется мало. Таким образом, у большинства пациентов обнаруживается сниженное соотношение CD4+- и CD8+-T- клеток, что приводит к инверсии нормального Т-клеточного индекса CD4:CD8. Присутствующие С04+-Т-клетки функционально полноценны, что подтверждается их способностью отвечать на стимуляцию in vitro. Для предотвращения развития РТПХ этим пациентам необходим донор костного мозга, подходящий по антигенам HLA.
Мутация, ответственная за СГЛ, располагается не в самих генах HLA II класса, а в одном из четырех генов, кодирующих регуляторные факторы, необходимые для транскрипции генов HLA II класса. Можно предположить, что лучшее понимание нарушений транскрипции при СГЛ может привести к разработке метода, позволяющего регулировать экспрессию HLA II класса. Он может применяться для предотвращения отторжения трансплантата при пересадке органов (например, почек, печени) у иммунокомпетентных пациентов.
Было выявлено лишь несколько пациентов с дефицитом экспрессии молекул HLA I класса; некоторые из них были обнаружены случайно. Это, несомненно, связано с тем, что не у всех лиц с дефицитом экспрессии молекул HLA I класса развивается клинически значимый иммунодефицит. Как и у пациентов с СГЛ с дефектом экспрессии молекул HLA II класса, у лиц с недостаточностью молекул HLA I класса мутация гена молекул HLA I класса отсутствует, но изменен ген, кодирующий белок-транспортер (ТАР). Как известно, белок-транспортер переносит пептиды, синтезированные в цитозоле, в эндоплазматический ретикулум, где они взаимодействуют и стабилизируют структуру молекул МНС I класса. При отсутствии продукта гена ТАР экспрессия молекул МНС I класса на клеточной поверхности очень низкая. По непонятным причинам у пациентов с клинически значимым дефектом чаще возникают рецидивирующие пневмонии, нежели ожидаемые вирусные инфекции.
Мутация ZAP-70. Пациенты с мутацией Т-клеточной тирозинкиназы ZAP-70, которая переводит сигнал, полученный от Т-клеточного рецептора, также имеют ТКИД-подобный фенотип.
Другие многосистемные расстройства
В дополнение к комбинированным иммунодефицитным заболеваниям, которые мы только что обсудили, некоторые полисистемные наследственные нарушения приводят к ТКИД-подобной клинической картине.
Синдром Вискотта-Олдрича. Синдром Вискотта-Олдрича — заболевание, сцепленное с хромосомой X, характеризующееся классической триадой симптомов: геморрагический диатез (тенденция к кровоточивости) вследствие тромбопитопении (низкого содержания тромбоцитов в крови) и малого размера тромбоцитов, повторяющиеся бактериальные инфекции и аллергические реакции (включая экзему, повышенный уровень IgE и пищевую аллергию). Кроме того, у этих пациентов наблюдается высокий риск развития онкопатологии, особенно опухолей лимфоидной ткани. Генетической основой этого заболевания является мутация гена хромосомы X, кодирующего белок синдрома Вискотта-Олдрича (Wiskott-Aldrich syndrome protein — WASP), который экспрессируется на всех стволовых гемопоэтических клетках. Современные данные позволяют предположить, что WASP взаимодействует с цитоскелетом, и в клетках пациентов с этим синдромом цитоскелет становится не способным видоизменяться в ответ на стимуляцию.
Иммунные дефекты вариабельны и затрагивают функции обеих популяций Т- и В-клеток; особенно снижено количество Т-клеток. Характерно, что у этих пациентов не возникает ответ на полисахаридные антигены. Лечение таких больных состоит в немедленном назначении антибиотиков и противовирусных препаратов при любой инфекции. После трансплантации костного мозга происходит восстановление функции Т- и В- клеток. Без терапии средняя продолжительность жизни пациентов составляет около 3 лет. С увеличением продолжительности жизни можно ожидать повышения заболеваемости онкологическими заболеваниями.
Атаксия-телеангиоэктазия. Другим полисистемным генетическим заболеванием является атаксия-телеангиоэктазия, при которой неврологические симптомы (шатающаяся походка или атаксия) и патологическое расширение сосудов (телеангиоэктазия) сопровождают повышенную чувствительность к инфекциям, лимфопению (низкий уровень лимфоцитов в периферической крови), гипоплазию тимуса и пониженные уровни IgA, IgE и иногда IgG. Иммунный дефект охватывает оба звена: клеточный и гуморальный (Т-зависимый и Т-независимый) иммунный ответы, особенно сильно поражая Т-зависимые участки лимфоидной ткани. В основе генетического дефекта этого синдрома лежит мутация гена, кодирующего белок (ataxia telangiectasia mutated — ATM), который участвует в процессе репарации повреждений клеточной ДНК после воздействия ионизирующего излучения или оксидативного взрыва. У больных атаксией-телеангиоэктазией нарушено развитие Т- и В-клеток. При нормальном развитии Т- и В-клетки проходят важные фазы интенсивной клеточной пролиферации, апоптоза и процессы рекомбинации ДНК, каждая из которых может быть нарушена под влиянием мутированного нефункционального белка ATM. Дети с атаксией-телеангиэктазией также имеют высокий риск развития онкологической патологии, особенно новообразований лимфоидной ткани. Это может быть связано с дефектом АТМ-зависимого восстановления ДНК или нарушением клеточного цикла после хромосомного повреждения. Заболевание ассоциировано с синдромом Блума и анемией Фанкони. Оба эти заболевания характеризуются одинотипным вариабельным иммунным дефицитом и чувствительностью к повреждению ДНК.
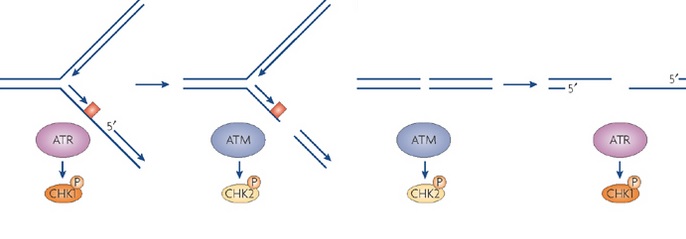
Таким образом, указанные причины, приводящие к тяжелым дефектам обоих звеньев иммунитета, клеточно-опосредованного и гуморального, различны. Они варьируют от мутаций ферментов, находящихся во всех клетках, что должно оказывать огромное воздействие на организм в целом, до мутаций, вовлекающих специфические сигнальные белки, экспрессированные в Т-клетках (мутация ZAP-70).
Для понимания природы синдромов, наблюдаемых у людей, и стадий нормального развития Т- и В-клеток, много информации можно получить, изучая модели на животных. Для изучения этой группы заболеваний первыми моделями были ТКИД-мутантные породы мышей, имеющие генетический дефект белка, восстанавливающего обрывы двухцепочечной ДНК. Впоследствии были выведены породы «нокаутных» мышей, моделирующих большую часть спонтанных генетических заболеваний человека, что оказало существенную помощь в исследованиях. Кроме того, ТКИД-мышей и «голых» мышей (обсуждается далее в этой главе) с ослабленной способностью к отторжению чужеродных тканей можно использовать в качестве «живой тест-пробирки» для изучения роста стволовых гемопоэтических клеток и опухолей человека.
Иммунодефицитные состояния, связанные с Т-клетками и клеточно-опосредованным иммунитетом
Пациенты с недостаточностью Т-клеточного звена восприимчивы к вирусной, грибковой и протозойной инфекциям Более того, поскольку Т-клетки необходимы для продукции В-клетками антител к Т-зависимым антигенам, у пациентов с дефицитом Т-клеточного звена также проявляются избирательные дефекты продукции антител. Следовательно, пациентов с Т-клеточной недостаточностью бывает трудно отличить от больных с ТКИД по клинической картине.
Врожденная аплазия тимуса (синдром Ди Джорджи). Синдром Ди Джорджи — это Т-клеточная недостаточность, при которой тимус, а также другие нелимфоидные органы развиваются аномально. Этот синдром возникает вследствие нарушения миграции клеток эмбрионального нервного гребешка в третий и четвертый глоточные карманы. В норме этот процесс происходит на 12-й неделе гестации. При синдроме Ди Джорджи развиваются аномалии сердца, лицевой области, тимуса и паращитовидных желез, что приводит к аплазии тимуса и гипопаратиреоидизму. Следствием аплазии тимуса является отсутствие зрелых Т-клеток и иммунодифицит. Моделью синдрома Ди Джорджи являются «голые» мыши; у этих животных не развиваются тимус и волосяные фолликулы.
Синдром Ди Джорджи не является наследственно обусловленным, но встречается спорадически как результат делении хромосомы 22ql 1. У новорожденных отмечают гипокальциемию (низкий уровень кальция) вследствие отсутствия паращитовидных желез и врожденные пороки сердца. Такие дети страдают от частых или хронических вирусных, бактериальных, грибковых и протозойных инфекций. У них отсутствуют зрелые Т-клетки на периферии (в крови, лимфатических узлах или селезенке) или их очень мало. Хотя В-клетки, плазматические клетки и уровни Ig в плазме могут быть нормальными, у многих пациентов не происходит увеличения ответа антител после иммунизации Т-зависимыми антигенами. Недостаток Т-клеток-хелперов, необходимых для переключения на другие изотипы иммуноглобулинов, приводит к отсутствию IgG и антител других изотипов после иммунизации. Однако, ответ IgM на Т-независимые антигены не страдает. Поскольку больные с синдромом Ди Джорджи имеют недостаточность Т-клеточного звена и не способны генерировать нормальный антительный ответ, их нельзя прививать живыми ослабленными вирусными вакцинами!
Изначально детей с синдромом Ди Джорджи лечили пересадкой эмбрионального тимуса, что приводило к появлению в организме хозяина Т-клеток в течение недели. Возраст эмбрионального тимуса, используемого для трансплантации, должен быть менее 14 недель гестации, что обеспечивает отсутствие РТПХ, которая может встречаться, если реципиенту с иммунными нарушениями (иммунокомпрометированному) были внесены зрелые донорские клетки тимуса. Донорский эмбриональный тимус обеспечивает наличие тимических эпителиальных клеток, которые позволяют нормальным лимфоидным клеткам-предшественникам реципиента развиться до Т-лимфоцитов. Хотя образовавшиеся Т-клетки нормальные, клеточно-опосредованный иммунитет и помощь в продукции антител восстанавливаются не полностью. Т-клетки реципиента «запоминают» как «свои» молекулы МНС клеток трансплантированного тимуса и иногда плохо взаимодействуют с АПК собственного организма на периферии. Поскольку эта лечебная стратегия не была успешной, то в настоящее время в основном проводят симптоматическую терапию. Некоторые пациенты имеют резидуальную тимическую ткань, которая обеспечивает хотя запоздалое и уменьшенное, но все-таки развитие Т-клеток. Дополнительной медицинской проблемой, связанной с этим синдромом, является врожденный порок сердца, что ухудшает в общем-то и без того плохой прогноз.
Т-клеточная недостаточность с нормальным количеством периферических Т-клеток
У определенного числа пациентов были обнаружены в большей степени функциональные, нежели количественные дефекты Т-клеток. Клинически они могут проявляться оппортунистическими инфекциями и удивительно высокой частотой аутоиммунных заболеваний. Исследование родословных в таких семьях выявляет аутосомно-рециссивный тип наследования. Исследования на молекулярном уровне демонстрируют гетерогенность причинно-значимых факторов, в том числе недостаточную экспрессию тирозинкиназы ZAP-70, CD3е.
ZAP-70 требуется для внутриклеточного преобразования сигнала после связывания с Т-клеточным рецептором. По неясным причинам пациенты с дефектом экспрессии ZAP-70 имеют клиническую картину ТКИД-подобного синдрома (с дефектом клеточно-опосредованного и гуморального иммунитета). Отсутствие Т-клеточной активности позволяет предположить, что ZAP-70 играет решающую роль в функционировании зрелых Т-клеток, однако причины воздействия на функции В-клеток не ясны. Кроме того, хотя количество периферических клеток крови, лимфатические узлы и тимус исходно нормальны, у пациентов с дефектом экспрессии ZAP-70 отсутствуют С08+-Т-клетки. Этот факт позволяет предположить, что ZAP-70 также требуется для дифференцировки CD8+-T- клеток в тимусе. Мутации цепей CD3 достаточно редки, и описано малое количество пациентов с таким дефектом. Модели на животных подтверждают, что пептидные цепи CD3 необходимы для нормальной передачи сигнала через Т-клеточный рецептор. Однако неясно, точно ли соответствуют такие мышиные модели тому дефекту, который имеется у описанных ранее пациентов.
Аутоиммунный лимфопролиферативный синдром
Аутоиммунный лимфопролиферативный синдром (АЛПС) — наследственное заболевание, характеризующееся массивной пролиферацией лимфоидной ткани с ранним развитием лимфомы. Оно может считаться аутоиммунным, поскольку такой генетический дефект приводит к системному аутоиммунному феномену и повышенной восприимчивости только к хроническим вирусным инфекциям. У пациентов повышается количество дважды негативных (CD4 CD8 ) Т-клеток и может развиться В-клеточная лимфома. Большинство больных АЛПС имеют мутацию гена, кодирующего белок Fas (CD95). Сигналы, проводимые через этот белок, в норме активируют апоптоз, или программируемую гибель клетки. Без активации апоптоза клетки, которые должны погибнуть, живут, а иммунные реакции, которые должны «выключиться», продолжаются. Большинство больных АЛПС имеют одну нормальную и одну мутантную молекулу Fas. Это позволяет предположить, что мутантная молекула каким-то образом мешает функционированию нормальной молекулы Fas. Некоторые больные АЛ ПС имеют дефекты других компонентов запуска апоптоза, таких как Fas-лиганд или каспаза 10.
Две породы мышей — lps и gld — обладают фенотипом, схожим с фенотипом больных АЛПС; lps- мыши имеют мутацию в гене Fas, а gld-мыши — в гене Fas-лиганда. В течение многих лет lps-мыши изучались как модель аутоиммунного заболевания, особенно СКВ, пока не был обнаружен дефект в их Fas-гене.
Хронический кожно-слизистый кандидоз
Хронический кожно-слизистый кандидоз — это плохо определяемая совокупность синдромов, характеризующихся инфицированием кожи и слизистых оболочек грибами рода Candida. Эти грибы обычно присутствуют на поверхности таких тканей, но не являются патогенным. У пациентов обычно не нарушен клеточно-опосредованный иммунитет к другим микроорганизмам, отличным от грибов рода Candida, а также нормальный опосредованный В-клетками иммунитет (продукция антител) в ответ на все микроорганизмы, включая Candida. Таким образом, имеется только избирательный дефект функционирования Т-клеток. Это заболевание поражает и мужчин, и женщин, особенно часто проявляется в детском возрасте, а в некоторых случаях может быть наследственным.
В-клеточные или иммуноглобулинассоциированные иммунные заболевания варьируют от заболеваний с дефектом развития В-клеток и полным отсутствием всех классов Ig до заболеваний, связанных с недостаточностью одного класса или подкласса Ig. Больные страдают от рецидивирующих или хронических инфекций, которые могут начаться в детстве (агаммаглобулинемия Брутона) или молодом возрасте. Для диагностики этих заболеваний необходимо узнать количество В-клеток и исследовать их функцию, провести иммуно-электрофоретическое и количественное определение классов и подклассов Ig.
Х-сцепленная младенческая агаммаглобулинемия
Х-сцепленную младенческую агаммаглобулинемию (ХСА) впервые описал в 1952 г. О. Брутон (О. Bruton). Это заболевание также называется агоммаглобулинемией Брутона. Такое расстройство встречается сравнительно редко (1 случай на 100000 чел.). Впервые оно проявляется в возрасте 5-6 мес, когда младенец теряет материнские IgG, полученные через плаценту. В этом возрасте у младенцев основными проявлениями являются тяжелые рецидивирующие бактериальные инфекции как результат тяжелого угнетения или, возможно, отсутствия иммуноглобулинов всех классов.
В основе главного дефекта лежит неспособность предшественников В-клеток, которые представлены в нормальном количестве, превращаться в зрелые В-клетки. Ген ВТК (Брутоновской тирозинкиназы), который подвергается мутациям при ХСА, в норме кодирует фермент тирозинкиназу, расположенную в цитозоле. Он, по-видимому, является незаменимым участником процесса преобразования сигнала, передаваемого от пре-В-клеточного рецептора, в развивающихся В-клетках. Без этого сигнала клеточное развитие не продолжается. Все зрелые В-клетки у лиц, получивших от матерей-носителей мутантный ген, образуются только благодаря наличию другой активной (немутантной) хромосомы X. Таким образом, ХСА является одним из нескольких описанных наследственно обусловленных иммунодефицитов, за которые ответственна мутация цитоплазматической тирозинкиназы. Мутации JAK3, вызывающие развитие одной из форм ТКИД, и ZAP-70, ответственные за форму Т-клеточной недостаточности, были описаны ранее.
Исследования крови, костного мозга, селезенки и лимфатических узлов у пациентов с ХСА выявляют почти полное отсутствие В-клеток и плазматических клеток, что объясняет низкие уровни Ig. Характерно, что у детей значительно недоразвиты миндалины. В-клетки, образующиеся в ограниченном количестве, обладают нормальной способностью к превращению в плазматические клетки. Младенцы с ХСА страдают от рецидивирующих:
-
бактериальных средних отитов;
-
бронхитов;
-
септицемии;
-
пневмонии;
-
артритов;
-
менингитов;
-
дерматитов.
Наиболее часто обнаруживаются микроорганизмы Hemophilus influenzae и Streptococcus pneumoniae. Часто пациенты также страдают от мальабсорбции из-за засилья в ЖКТ Giardia lamblia. При этом такие больные также чувствительны к вирусной инфекции, которая проникает через ЖКТ, например эховирусам и вирусу полиомиелита. Инфекции плохо поддаются только одним антибиотикам, поэтому лечение состоит из периодических инъекций внутривенного иммуноглобулина (ВВИТ), содержащего большое количество IgG. Хотя эта пассивная иммунизация поддерживает некоторых пациентов в течение 20-30 лет, прогноз дается с осторожностью, поскольку из-за частого рецидивирования инфекций высока вероятность развития хронических легочных заболеваний.
Транзиторная гипогаммаглобулинемия
У младенцев в возрасте 5-6 мес. пассивно перенесенные материнские IgG исчезают, а продукция иммуноглобулинов в организме начинает возрастать. У недоношенных младенцев может быть транзиторная недостаточность IgG, поскольку они еще не способны синтезировать иммуноглобулины. Время от времени у младенцев, рожденных в срок, также может отмечаться снижение уровня IgG даже при нормальных уровнях IgM и IgA. Это состояние возникает вследствие снижения количества Т-хелперов и нарушения их функции. Транзиторная гипогаммаглобулинемия может сохраняться от нескольких месяцев до 2 лет. Это заболевание не связано с полом и его можно отличить от Х-сцепленного заболевания по наличию нормального количества В-клеток в кровотоке. Хотя обычно в лечении нет необходимости, нужно выявлять таких младенцев, поскольку в этот период не следует проводить иммунизацию.
Общая вариабельная иммунная недостаточность
У пациентов с общей вариабельной иммунной недостаточностью (ОВИН) отмечаются значительно сниженные уровни сывороточных IgG и IgA, при этом уровень IgM нормальный или сниженный, а количество В-клеток в периферической крови нормальное или сниженное. Причина заболевания, которое поражает и мужчин, и женщин, не совсем понятна, а возможно, и не является одинаковой во всех случаях. Начало заболевания может отмечаться в любом возрасте, но выделяют два пика в 1-5 и 15-20 лет. У заболевших отмечают повторные респираторные и кишечные инфекции, вызванные пиогенными бактериями, и аутоиммунные заболевания, такие как гемолитическая анемия, тромбоцитопения и СКВ, которые ассоциируются с аутоантителами. У многих также наблюдаются расстройства клеточно-опосредованного иммунитета. В долгосрочной перспективе у таких пациентов повышена частота онкологических заболеваний, в частности лимфом и рака желудка.
Общая вариабельная иммунная недостаточность характеризуется нарушением созревания В-клеток в клетки, секретирующие антитела. Этот дефект может быть обусловлен неспособностью В-клеток пролиферировать в ответ на антиген или нормальной пролиферацией В-клеток без секреции IgM, или секрецией IgM без переключения изотипа на IgG или IgA (вследствие внутреннего повреждения В- или Т-клеток), или нарушением гликозилирования тяжелых цепей IgG. В большинстве случаев расстройство проявляется в результате угнетения синтеза и секреции иммуноглобулинов. Это заболевание является семейным или возникает спорадически, при этом факторы внешней среды, вызывающие заболевание, неизвестны.
Лечение зависит от степени тяжести. При тяжелых формах болезни с множеством повторных или хронических инфекций назначается лечение внутривенным иммуноглобулином. У больных, получающих лечение, как правило, нормальная продолжительность жизни. У матерей с ОВИН беременность протекает нормально, но, безусловно, они не передают материнские IgG плоду.
Избирательные дефициты иммуноглобулинов
Несколько синдромов обусловлены селективными дефицитами одного класса или подкласса иммуноглобулинов. Некоторые из них сопровождаются компенсаторным увеличением уровней других изотипов антител, как, например, увеличение концентрации IgM в случаях дефицита IgG или IgA.
Недостаточность IgA является наиболее распространенным иммунодефипитным расстройством на Западе, его частота ориентировочно составляет один случай на 800 чел.. Причина этого расстройства неизвестна, но, похоже, связана со снижением высвобождения IgA из В-лимфоцитов. Дефицит IgA также может возникать и транзиторно, в качестве побочного эффекта действия лекарственных препаратов. Пациенты могут страдать от рецидивирующих инфекций верхних и нижних дыхательных путей вирусной или бактериальной этиологии, целиакии (нарушенной абсорбции в кишечнике) или не иметь никаких симптомов.

Лечение пациентов, у которых все же отмечаются проявления заболевания, состоит в назначении антибиотиков широкого спектра действия. Терапия сывороточным иммуноглобулином не применяется, поскольку промышленные препараты содержат низкие концентрации IgA и потому что IgA, введенный парентерально, не достигает участков секреторной иммунной системы, где обычно является защитным иммуноглобулином. Более того, у пациентов может развиваться гуморальный ответ (обычно IgG или IgE) к IgA из перелитой иммунной сыворотки, что вызывает реакции гиперчувствительности. Тем не менее, прогноз при селективном дефиците IgA в основном хороший, и большинство пациентов живут нормально.
Отмечаются селективные дефициты и других изотипов иммуноглобулинов. Примером является недостаточность IgM, редкое расстройство, при котором пациенты переносят повторные и тяжелые инфекции, вызванные микроорганизмами, имеющими полисахаридные капсулы, такие как пневмококки или Haemophilus influenzae. Описаны избирательные дефициты подклассов IgG, но они чрезвычайно редки.
Расстройства взаимодействия Т-В-лимфоцитов
Существуют по меньшей мере два заболевания, при которых линии Т- и В-клеток созревают нормально, но взаимодействия между их представителями нарушены. Хотя причиной обоих этих состояний являются нарушения в Т-клетках, преобладающие клинические проявления связаны с В-клетками или гуморальным иммунным ответом. Этими заболеваниями являются гипep-IgM-син- дром и Х-сцепленная лимфопролиферативная болезнь.
-
Гипер-IgM-синдром.
Х-сцепленный гипep-IgM-синдром (ХГИМ) проявляется в возрасте 1-2 лет рецидивирующими респираторными инфекциями и очень низкими уровнями сывороточных IgG, IgA и IgE в сочетании с нормальным или повышенным уровнем IgM. Количество В-клеток у таких пациентов нормальное, in vitro они полноценно функционируют и способны к переключению изотипа при соответствующей стимуляции. Т-клетки также присутствуют в обычном количестве, нормально распределены по субпопуляциям и отвечают пролиферацией на митогены. Мутация в гене CD40L4 расположенном на хромосоме X, приводит к отсутствию лиганда CD40 (CD154) на Тн-клетках. При этом CD40L связывается с CD40, экспрессируемым на В-клетках. Это взаимодействие предотвращает апоптоз В-клеток и представляется важным, если не необходимым, для переключения изотипа. При ХГИМ также не происходит привлечения В-клеток в фолликулы, что приводит к отсутствию сформированных зародышевых центров. У мальчиков с таким расстройством также наблюдаются небольшие изменения в функциях Т-клеток и частичный блок дифференцировки нейтрофилов и активации макрофагов. Это может объяснять их подверженность оппортунистическим инфекциям, в частности пневмониям, вызванным Pneumocystis carinii, и прогноз у них хуже, чем у пациентов с ХСА.
У второй группы пациентов с такими же клиническими проявлениями, как и при ХГИМ, но с аутосомно-рецессивным типом наследования, возможно, имеется дефект В-клеток в молекуле CD40 У пациентов третьей группы с дефектом взаимодействия CD40 с модулятором транскрипционного фактора NF-кВ отмечается Х-сцепленный механизм наследования. Как это часто бывает при генетических нарушениях, вовлекающих такого типа регуляторные молекулы межклеточных взаимодействий, у заболевших детей также наблюдаются патологические изменения в клетках, не принадлежащих иммунной системе. Например, такие регуляторные молекулы используют множество типов клеток.
-
Х-сцепленное лимфопролиферативное заболевание (синдром Дункан).
Впервые Х-сцепленное лимфопролиферативное (ХСЛ) заболевание было описано у шести родственников по материнской линии мужского пола в семье Дункан, поэтому такое название стало нарицательным. Основным компонентом комплексного дефекта, определяющего развитие этого редкого заболевания, признана неспособность Т-клеток регулировать рост В-клеток. До контакта с вирусом Эпштейн-Барр такие пациенты клинически здоровы, у них отмечается нормальное количество Т- и В-клеток. Однако при воздействии вируса у них развивается тяжелая форма инфекционного мононуклеоза, которая может быть летальной. В том случае, если пациент выживает после перенесенной инфекции, у него часто развивается злокачественная лимфома или дизгаммаглобулинемия. Лимфома или дефицит иммуноглобулинов могут возникнуть у них и без предварительного контакта с вирусом Эпштейн-Барр. Среди лимфом преимущественно отмечаются агрессивные В-клеточные лимфомы вне лимфатических узлов (экстранодально), в частности в ЖКТ. Наиболее частым типом является лимфома Беркитта. Несмотря на то, что тип развивающихся лимфом такой же, как и у пациентов с другими нарушениями контроля индуцируемой вирусом Эпштейн-Барр пролиферации В-клеток, вызванной дефектом Т-клеток (например, пациентов со СПИД или пациентов, получающих иммуносупрессивную терапию после трансплантации), частота развития лимфом при ХСЛ значительно выше. Прогноз крайне неблагоприятный.
Дисфункции фагоцитирующих клеток
Фагоцитирующие клетки — полиморфноядерные лейкоциты и макрофаги/моноциты — играют очень важную роль, как во врожденном, так и в приобретенном иммунитете, действуя против патогенов самостоятельно или вместе с лимфоцитами. Наследственные синдромы недостаточности фагоцитирующих клеток помогли идентифицировать множество молекул, необходимых на всех стадиях в действиях фагоцита, необходимых для уничтожения возбудителя. К этим стадиям и связанным с ними дефицитам относятся:
-
миграция и адгезия фагоцитирующих клеток (недостаточность лейкоцитарной адгезии);
-
фагоцитоз и сплавление с лизосомами (синдром Чедиака-Хигаси);
-
окислительный взрыв для уничтожения возбудителя (хроническая гранулематозная болезнь).
Фагоцитарная дисфункция может быть и вторичной, вызванной внешними факторами, такими как лекарственные препараты и системные заболевания (например, сахарный диабет), или дефектами других звеньев иммунной системы.
-
Недостаточность лейкоцитарной адгезии.
Чтобы лейкоциты прибыли к месту инфекции в тканях, клеткам, прежде всего, необходимо покинуть кровоток. Этот процесс состоит из нескольких стадий. На первой лейкоциты начинают медленно перекатываться вдоль эндотелия благодаря взаимодействиям селектинов на эндотелиальных клетках и лигандов-селектинов на лейкоцитах. Затем под действием хемоаттрактантов клетка прекращает перемещение (прокатывание). Такая клетка прикрепляется более плотно, а затем начинает миграцию через эндотелий. Для осуществления последней стадии требуется взаимодействие интегринов на лейкоцитах со своими лигандами на эндотелиальных клетках.
Недостаточность лейкоцитарной адгезии (НЛА) — это группа расстройств, при которых прерывается взаимодействие лейкоцитов с эндотелием сосудов. Выделяют НЛА I — аутосомно-рецессивное заболевание, ген которого картирован на хромосоме 21. У пациентов выявляют дефект в р-субъединице интегриновых молекул, который препятствует их экспрессии. При этом р-субъединица является общей для трех интегринов, экспрессируемых на гранулоцитах, моноцитах и лимфоцитах — LFA-1 (CD1 la/CD18), Mac-1 (CD1 lb/CD 18) и р150,95 (CD1 lc/CD18) соответственно. В результате этого нарушается адгезия и миграция всех типов лейкоцитов. Пациенты с НЛА I страдают рецидивирующими бактериальными инфекциями мягких тканей, при которых у них повышается количество лейкоцитов, но не образуется гной или отсутствует эффективное заживление ран. Функции лимфоцитов также нарушены из-за отсутствия экспрессии LFA-1. Отличительной чертой новорожденных с НЛА I является позднее отпадение остатка пуповины.
У пациентов с НЛА II наблюдается дефект селектиновых лигандов, поэтому их клетки не могут прокатываться вдоль эндотелиальной поверхности (первый шаг миграции). Дефект, определяющий развитие НЛА II, связан с метаболизмом фукозы, что приводит к отсутствию фукозилированных лигандов, с которыми могли бы связываться селектины. Симптомы иммунодефицита при этом расстройстве менее выражены. Кроме них дефект метаболизма фукозы приводит к другим порокам развития. Как и при НЛА I, характерно отсутствие или незначительное образование гноя; у таких детей не выражены классические клинические признаки воспаления при тяжелых инфекциях.
-
Синдром Чедиака-Хигаси.
Синдром Чедиака-Хигаси является аутосомно- рецессивным заболеванием, которое характеризуется аномальными гигантскими гранулами и органеллами в клетках. В основном поражаются лизосомы и меланосомы, что приводит к дефектам пигментации и нарушениям функционирования нейтрофилов, NK-клеток и тромбоцитов, а также неврологическим аномалиям. У нейтрофилов снижена способность к внутриклеточному уничтожению организмов, что является результатом как дефектной дегрануляции, так и нарушения сплавления лизосом с фагосомами. Со временем у пациентов формируются массивные инфильтраты из лимфоцитов и макрофагов в печени, селезенке и лимфатических узлах. Пиогенные микроорганизмы, такие как стрептококки и стафилококки, вызывают рецидивирующие, иногда летальные инфекционные заболевания. Прогноз неблагоприятный.

-
Хроническая гранулематозная болезнь.
При хронической гранулематозной болезни (ХГБ) нарушается последняя стадия уничтожения поглощенных организмов. При этом внутриклеточная персистенция организмов приводит к формированию гранулем. У нормальных индивидуумов активированные нейтрофилы и мононуклеарные фагоциты уничтожают микроорганизмы посредством окислительного взрыва, при котором потребляется кислород и выделяются перекись водорода и свободные супероксидные радикалы. Мутации в любой из субъединиц фермента, катализирующего этот взрыв — НАДФ- оксидазы — могут привести к ХГБ. Наиболее распространенная форма ХГБ развивается из-за мутации в одной из субъединиц, связанных с мембраной, gp91phox, которая кодируется геном CYBB, расположенным на хромосоме X. Таким образом, у большинства пациентов отмечается Х-сцепленный рецессивный тип наследования. Другие субъединицы НАДФ-оксидазы кодируются аутосомными генами. У пациентов с ХГБ, вызванной мутациями в таких аутосомных субъединицах, отмечается аутосомно-рецессивный тип наследования. В основном у них имеются мутации в одной из двух цитозольных субъединиц фермента, p47phox или p67phox.
Симптомы заболевания появляются в первые 2 года жизни. У пациентов наблюдается повышенная восприимчивость к инфицированию микроорганизмами, в обычных условиях обладающих низкой вирулентностью, таким как Staphylococcus aureus, Serratia marcescens и Aspergillus. Сопутствующими аномалиями являются лимфаденопатия (увеличение размеров лимфатических узлов) и гепатоспленомегалия (увеличение размеров печени и селезенки), связанные с хроническими и острыми инфекциями. Лечение состоит в агрессивной иммунизации и терапии антибиотиками широкого спектра действия, противогрибковыми препаратами и интерфероном-у.
Кроме ХГБ к снижению способности уничтожать внутриклеточные организмы приводят расстройства, при которых наблюдается снижение или отсутствие активности ферментов, участвующих в процессе фагоцитоза. К ним относятся глюкоза-6-фосфатдегидрогеназа, миелопероксидаза и щелочная фосфатаза.
Недостаточность рецептора интерферона-у
Мутация в гене IFNyRl приводит к неспособности моноцитов отвечать на IFNy секрецией фактора некроза опухоли a (TNFa). Пациенты с этой мутацией избирательно восприимчивы к слабо патогенным микобактериям, что подтверждает значение IFNy в контроле над микобактериальными инфекциями. Эта избирательность также свидетельствует, что другие активности IFNy у лиц с этим расстройством компенсируются. Иммунизация БЦЖ, распространенная в некоторых частях света, может быть опасной для пациентов с этим дефектом.
Недостаточность NK-клеток
О недостаточности натуральных киллеров у людей известно очень мало; было описано лишь несколько случаев этого заболевания. Исследования на животных позволяют предположить, что недостаточность NK-клеток нарушает отторжение аллотрансплантатов и связана с повышенной восприимчивостью к вирусным заболеваниям и увеличенной частотой метастазирования опухолей. Дефекты NK-клеток наблюдаются при тяжелых комбинированных иммунодефицитных расстройствах, при некоторых расстройствах функций Т-клеток и фагоцитирующих клеток, а также при Х-сцепленном лимфопролиферативном синдроме.
Заболевания, вызванные нарушением системы комплемента
Комплемент необходим для опсонизации и уничтожения бактерий и поврежденных клеток, хемотаксиса и активации В-клеток. Компоненты комплемента также участвуют в уничтожении комплексов антиген-антитело, предотвращая депонирование иммунных комплексов и последующие заболевания. Недостаточность комплемента может наследоваться как аутосомный признак, при этом у гетерозиготных людей концентрация определенного компонента комплемента будет составлять половину от нормальной. Для большинства компонентов этого достаточно, чтобы предотвратить клинические проявления заболевания. Период полураспада активированных компонентов комплемента также обычно строго контролируется ингибиторами, которые разрушают продукты активации или диссоциируют комплексы.
Недостаточность компонентов комплемента ранней фазы активации
Компоненты комплемента ранней фазы активации особенно важны для образования опсонина СЗb. У пациентов с дефицитами С1, 4 или 2 по классическому пути активации или собственно дефицитом СЗ отмечаются более частые случаи инфицирования инкапсулированными организмами (Streptococcus pneumoniae, Streptococcus pyogenes, Haemophilus influenzae) и ревматические заболевания из-за нарушенного клиренса иммунных комплексов вследствие недостаточного образования СЗb. Наиболее заметным последствием этого являются частые аутоиммунные заболевания. Действительно, СКВ является одним из наиболее распространенных проявлений дефицита некоторых компонентов комплемента. У таких пациентов СКВ отличается очень ранним началом заболевания и большей тяжестью по сравнению с СКВ, не связанной с дефицитом комплемента. Кроме того, такая СКВ может развиваться в отсутствие антител, часто обнаруживаемых в других случаях. Недостаточность маннозосвязывающего лектина, который фиксируется на поверхности микробов без участия антител и запускает классический путь активации комплемента, также приводит к повышенному риску бактериальных инфекций и волчанкоподобным симптомам. Поскольку все пути активации комплемента — классический, лектиновый и альтернативный — требуют активации СЗ, недостаточность самого СЗ связана с наиболее тяжелыми симптомами, в частности осложненным течением инфекционных заболеваний.
Недостаточность компонентов комплемента поздней фазы активации
При недостаточности компонентов комплемента поздней фазы активации (С5-С9) нарушается образование мембраноатакующего комплекса (МАК). Этот комплекс является непосредственным разрушителем грамотрицательных бактерий, в частности Neisseria meningitidis, и первой линией защиты от них.
Нарушенный контроль за компонентами комплемента
Наследственный ангионевротический отек. При наследственном ангионевротическом отеке у пациентов отсутствует действующий ингибитор C1-эстеразы. Если этот ингибитор отсутствует, действие С1 на С4, С2 и новую систему не контролируется, что приводит к образованию больших количеств вазоактивных пептидов. Эти пептиды вызывают повышение проницаемости кровеносных сосудов. У пациентов отмечаются локализованные отеки, которые могут угрожать жизни, если развиваются в области гортани, перекрывая дыхательные пути. Лечение заключается в устранении провоцирующих факторов, к которым обычно относятся травмы, и проведении инфузий ингибитора эстеразы С1.
Недостаточность белков, связанных с гликозилированным фосфатидилинозитолом. Семейство белков, заякоренных ГФИ, экспрессируется на мембранах эритроцитов, лимфоцитов, гранулоцитов, эндотелиальных клеток (клеток, выстилающих кровеносные сосуды) и эпителиальных клеток. Эти белки, к которым относятся фактор, ускоряющий диссоциацию (ФУД, или CD55), и CD59, защищают клетки от спонтанного лизиса комплементом. Если ингибиторы отсутствуют на поверхности клетки, гранулоциты, тромбоциты и особенно эритроциты восприимчивы к спонтанному лизису комплементом. Существует лишь несколько семей с наследуемыми мутациями ФУД, CD59 или всех белков ГФИ. У таких пациентов отмечаются симптомы тяжелой анемии, тромбозы и хронические инфекции.
Приобретенная форма этого заболевания, называемая пароксизмальной ночной гемоглобинурией (ПНГ), встречается намного чаще. При ПНГ у пациентов отсутствует фермент, необходимый для продукции всех белков, заякоренных ГФИ. Это происходит вследствие приобретенной соматической мутации в ранней миелоидной клетке-предшественнике. Поражаются три нелимфоидные линии:
-
гранулоциты;
-
тромбоциты;
-
эритроциты.
У большинства пациентов стволовые клетки с этой мутацией приобретают и дополнительные мутации, а затем начинают доминировать над нормальными клетками в костном мозге и прекращают созревание, что приводит к острому миелоидному лейкозу. При хроническом течении ПНГ развивается внутрисосудистый гемолиз, особенно заметный по ночам в почках, где кислая окружающая среда запускает активацию комплемента по альтернативному пути. Это клиническое проявление и отражается в названии заболевания.
Вторичные иммунодефицитные заболевания
Вторичные иммунодефицитные заболевания являются последствиями других заболеваний. По- прежнему самой распространенной причиной иммунодефицитных расстройств по всему миру является неправильное питание (или его недостаточность). В развитых странах более часто встречается ятрогенный иммунодефицит, т.е. вызванный лекарственной терапией, в частности в результате использования химиотерапевтических агентов при лечении онкологических заболеваний или контролируемой иммуносупрессией в случаях пересадки органов или аутоиммунных заболеваний. Вторичные иммунодефициты также развиваются при аутоиммунных заболеваниях (не как следствие лечения) или после выздоровления от тяжелых бактериальных инфекций. Злокачественные новообразования иммунной системы также часто подавляют нормальные (немалигнизированные) компоненты, что проявляется у таких пациентов повышенной чувствительностью к инфекционным заболеваниям.








- Аллергия
- Ангиология
- Болезни глаз
- Венерология
- Гастроэнтерология
- Гинекология
- Дерматология
- Здоровое питание
- Инфекционные болезни
- Кардиология
- Косметология
- Лекарства
- Лекарственные растения
- ЛОР-заболевания
- Мужское здоровье
- Неврология
- Неотложная помощь
- Новости
- Онкология
- Ортопедия
- Паразитология
- Педиатрия
- Пульмонология
- Расшифровка анализов
- Симптомы
- Системные заболевания
- Стоматология
- Травматология
- Урология
- Хирургия
- Эндокринология
- Нужно знать
- Еда
- Профессиональные заболевания
Комментарии