
Амилоидоз: причины, симптомы, диагностика, лечение, фото
Амилоидоз представляет собой системное заболевание, характеризующееся внеклеточным депонированием различных нерастворимых белков. Эти белки могут накапливаться локально, являясь причиной появления соответствующих симптомов, либо иметь широкое распространение, включая многие органы и ткани, являясь причиной значительных системных расстройств и поражений. Амилоидоз может быть первичным и вторичным, являясь следствием развития различных инфекций, воспалительных процессов или злокачественных новообразований. Редко амилоидоз возникает в результате проявления нескольких наследственных метаболических дефектов. Диагностика основывается на результатах биопсии вовлеченных в процесс тканей. Лечение зависит от типа амилоидоза.
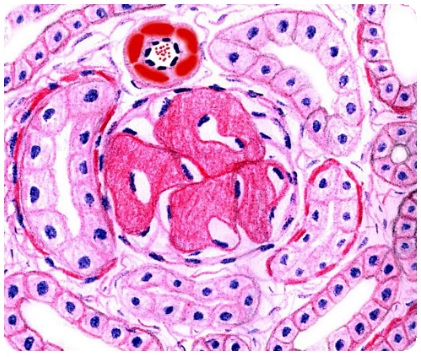
Этиология, патофизиология и классификация
Амилоидные отложения могут формироваться, по крайней мере, из 18 различных белков, включая фрагменты иммуноглобулина. Амилоидные отложения являются метаболически инертными, но способны физически изменять внутреннюю структуру органа и его функцию. Окраска препаратов положительная при окрашивании Конго красным, розовая — при окрашивании гематоксилином и эозином и имеет яблочно-зеленый цвет свечения в поляризованном свете после окраски Конго красным. Амилоидные отложения имеют волокнистую, обычно жесткую и неразветвленную ультраструктуру. Они формируют складчатый слой, который может быть виден при дифракции поляризованных лучей. В дополнение к фибриллярному белковому амилоиду эти отложения также содержат сывороточный амилоидный Р-компонент и гликозаминогликаны. При внимательном исследовании вовлеченных в процесс органов, в перечень заболеваний включают ревматоидный артрит (РА), болезнь Крона, наследственную приходящую лихорадку. Воспалительные цитокины (например, IL-1, фактор некроза опухолей, IL-6), которые выделяются при этих заболеваниях, являются причиной увеличения продукции белка-предшественника SAA клетками печени, циркулирующего в плазме крови.
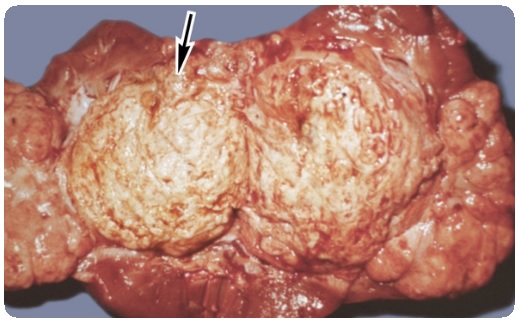
При АА-амилоидозе существует предрасположенность к поражению селезенки, печени, почек, надпочечников и лимфатических узлов. Печень, селезенка и почки часто увеличены в размерах, твердые и резиноподобной консистенции. Вовлечение в патологический процесс сердца и периферической или автономной нервной системы является редким явлением. Тем не менее, нет ни одной системы органов, которая в конечном итоге оставалась бы незатронутой болезнью, а вовлечение в патологический процесс сосудов может носить широко распространенный характер.
-
Семейный амилоидоз.
Наследственная форма амилоидоза является результатом накопления мутировавшей формы белка плазмы [наиболее часто транстиретина (TTR), отсюда — ATTR]. Раньше всего все белковые аномалии появляются в печени. На сегодняшний день установлено около 80 мутаций гена TTR. Все они передаются по аутосомно-доминантному пути наследования. Возраст манифестации симптомов является весьма вариабельным и варьирует от подросткового возраста до 70 лет. ATTR-амилоидоз вызывает периферическую сенсорную и моторную нейропатию, и часто автономную нейропатию. Характерным клиническим признаком при этом является развивающийся синдром карпального туннеля. Позднее, стечением болезни, в патологический процесс вовлекаются сердечно-сосудистая и мочевыводящая системы. Может также развиваться различная патология стекловидного тела.
Другие наследственные амилоидозы развиваются крайне редко и являются результатом мутаций других физиологических белков, включая аполипопротеин A-I, лизоцим, фибриноген, гельсолин (gelsolin), А р-протеин и цистатин (cystatin) С. Эти разновидности амилоидоза имеют множество системных и локальных эффектов.
-
Микроглобулиновый (диализ-ассоциированный) амилоидоз.
Эта форма встречается у пациентов с тяжелой хронической почечной недостаточностью, находящихся на плановом гемодиализе или перитонеальном диализе длительное время, обычно более 8 лет. Отложения амилоида включают 2-микроглобулин, компонент класса 1 главного комплекса гистосовместимости, который в норме метаболизируется почками, но не может быть выведен через диализные мембраны. Скопления амилоида предпочтительно откладываются вокруг костей и суставов, и в карпальном туннеле и обнаруживаются в ЖКТ и других органах.
-
Протеиновый амилоидоз.
Эта разновидность встречается у пациентов с болезнью Альцгеймера. Хотя окончательная роль амилоидных отложений до конца не ясна, невритические бляшки, образующиеся при болезни Альцгеймера, содержат амилоидные отложения, состоящие из белка-предшественника амилоида фрагмента протеина (трансмембранного гликопротеина). Этот фрагмент протеина иногда находится в комплексной связи с аполипопротеином Е. Внутренняя сторона бляшек — не волокнистая — сформирована из протеина, перемешанного с волокнистыми амилоидными формами.
Отложения протеинового амилоида могут также встречаться вокруг церебральных кровеносных сосудов, эти отложения, как полагают, являются причиной негипертонических церебральных кровоизлияний (церебральная амилоидная ангиопатия). Эта ангиопатия может встречаться спорадически или как наследственно обусловленный синдром (наследственное кровоизлияние в мозг голландского типа).
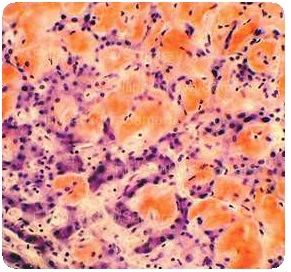
Симптомы и признаки
Клиническая симптоматика является неспецифической и связана с органом или системой органов, вовлеченных в патологический процесс. Симптомы при АА-амилоидозе часто завуалированы симптоматикой фонового заболевания, на фоне которого развился амилоидоз.
Когда в патологический процесс оказываются вовлеченными почки, то на первый план в клинической картине как наиболее раннее и выраженное проявление выступает нефротический синдром. Вначале может иметь место лишь небольшая протеинурия, позднее развивается комплекс характерных клинических симптомов: анасарка, гипопротеинемия и массивная протеинурия.
Вовлечение в процесс печени сопровождается безболезненной гепатомегалией, которая может быть довольно значительная (масса печени увеличивается до > 7 кг). За исключением редко наблюдаемого увеличения щелочной фосфатазы, печеночные ферменты остаются в пределах нормальных значений.
Желтуха развивается редко. Также редко развивается портальная гипертензия, с расширением вен пищевода и асцитом.
Поражение сердца заключается в развитии ограниченной кардиомиопатии, в конечном итоге приводящей к хронической сердечной недостаточности. Могут иметь место кардиомегалия и различной степени нарушения сердечной проводимости или аритмии.
Периферическая нейропатия, проявляющаяся парестезиями пальцев рук и ног, является характерной клинической симптоматикой, говорящей о манифестации AL- или ATTR-амилоидоза. Развивающаяся автономная нейропатия может быть причиной возникновения ортостатической гипотензии, эректильной дисфункции, нарушения потоотделения, расстройства желудочно-кишечной моторики.
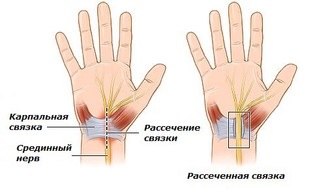
Ревматологическая симптоматика у пациентов с А р2-микроглобулиновым амилоидозом включает синдром карпального туннеля и хронический болевой синдром в области плеча, запястья и пальцев. Могут иметь место патологические переломы, особенно плечевой и бедренной костей.
Отложение амилоида в ЖКТ может стать причиной нарушения его моторики, особенно пищевода и тонкого, и толстого кишечника. Атония желудка, мальабсорбция, кровотечения или псевдонепроходимость могут также иметь место.
Характерным симптомом является макроглоссия.
Неподвижный, симметричный, плотный по консистенции зоб, такой же, как при тиреоидите Хашимото, может быть результатом амилоидоза щитовидной железы. Амилоидоз легких (в основном AL-амилоидоз) может характеризоваться образованием фокальных пульмонарных узлов, поражением трахеобронхиального дерева или диффузными альвеолярными отложениями. В некоторых случаях наследственного амилоидоза развивается помутнение стекловидного тела из-за отложения амилоида и билатеральная фестончатость краев зрачка.
Диагноз
Диагноз амилоидоза ставится клинически, но точное подтверждение его возможно только при проведении биопсии. Мягкая аспирация подкожного жира с поверхности передней брюшной стенки и биопсия слизистой прямой кишки являются наиболее результативными методами диагностики. Другими, широко используемыми местами забора биопсийного материала являются:
-
десна;
-
кожа;
-
нервная ткань;
-
почки и печень.
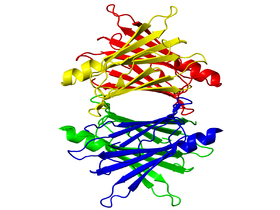
Мазки, взятые из образцов ткани, окрашиваются Конго красным и исследуются с помощью поляризованной микроскопии на наличие характерного двойного лучепреломления. Меченный изотопами сывороточный Р-компонент амилоида (в котором сам белок представлен пятиугольной структурой амилоида) может быть использован при сцинтиграфическом исследовании для подтверждения диагноза.
Прогноз
Прогноз зависит от типа амилоидоза и вовлеченных в процесс систем органов. AL-амилоидоз с миеломной болезнью имеет наихудший прогноз: как правило, наблюдается летальный исход в течение года. Нелеченый ATTR-амилоидоз также заканчивается фатально спустя 10-15 лет. При остальных формах семейного амилоидоза прогноз различен. В общем, поражение почек и сердца у пациентов с любым типом амилоидоза является весьма серьезной патологией.
Прогноз при АА-амилоидозе зависит от успеха лечения фонового заболевания, хотя довольно редко у пациентов наблюдается спонтанная регрессия амилоидных отложений без такого лечения.
Лечение
Лечение в основном симптоматическое, хотя терапия фоновой патологии может иногда приостанавливать развитие амилоидоза. У пациентов с амилоидозом почек длительное выживание после проведенной трансплантации почек сравнимо с таковым при другой почечной патологии. Хотя смертность у них в первые несколько лет выше. В конечном счете, наблюдается возвратное течение амилоидоза в донорской почке после трансплантации, хотя у некоторых пациентов наблюдается очень хорошее самочувствие и их выживаемость после операции приближается к 10 годам. У пациентов с амилоидозом и выраженным поражением сердца успешно проводится пересадка сердца.
Химиотерапия назначается пациентам с AL-амилоидозом. В протоколе лечения, как правило, используют мелфалан 0,075 мг/кг внутрь 2 раза в день и преднизолон 0,2 мг/кг внутрь 4 раза в день. Высокие дозы мелфалана успешно назначаются в случае пересадки костного мозга или стволовых клеток, чем достигается в некоторых случаях на короткий период положительный терапевтический эффект.
У пациентов с ATTR-амилоидозом весьма эффективна трансплантация печени, в результате чего полностью избавляются от органа — источника синтеза мутантного белка.
При АА-амилоидозе со средиземноморской семейной лихорадкой эффективен колхицин 0,6 мг/кг 1 или 2 раза в день внутрь. У пациентов с АА-амилоидозом и сопутствующими инфекциями последние следует активно лечить. Лечение амилоидоза, в основе развития которого лежат злокачественные новообразования (например, почечно-клеточная карцинома), направлено на лечение рака.








- Аллергия
- Ангиология
- Болезни глаз
- Венерология
- Гастроэнтерология
- Гинекология
- Дерматология
- Здоровое питание
- Инфекционные болезни
- Кардиология
- Косметология
- Лекарства
- Лекарственные растения
- ЛОР-заболевания
- Мужское здоровье
- Неврология
- Неотложная помощь
- Новости
- Онкология
- Ортопедия
- Паразитология
- Педиатрия
- Пульмонология
- Расшифровка анализов
- Симптомы
- Системные заболевания
- Стоматология
- Травматология
- Урология
- Хирургия
- Эндокринология
- Нужно знать
- Еда
- Профессиональные заболевания
Комментарии