
Вторичная надпочечниковая недостаточность. Врожденная гиперплазия коры надпочечников
Этиология
-
Острая отмена кортикостероидной терапии.
Вторичная надпочечниковая недостаточность чаще всего развивается в тех случаях, когда после длительного применения высоких доз активных глюкокортикоидов (что приводит к угнетению ГГНС) их внезапно отменяют или слишком быстро снижают их дозу. Такая опасность существует у больных лейкозом, астмой (особенно при переходе с пероральных на ингаляционные препараты кортикостероидов), диффузными болезнями соединительной ткани или другой аутоиммунной патологией, а также у тех, кто перенес трансплантацию какого-либо органа или нейрохирургическую операцию. Как долго можно вводить глюкокортикоиды и в какой дозе, чтобы избежать угнетения гипоталамуса и гипофиза, неизвестно, но считается, что при недельном их применении в количестве, на порядок превышающем физиологическую секрецию кортизола, препараты можно отменять сразу, а не постепенно. С другой стороны, при приеме детьми с лейкозом больших доз дексаметазона для восстановления функции надпочечников после отмены препарата требуется больше 2 мес. Надпочечниковая недостаточность у таких больных часто проявляется при последующих инфекциях или хирургических вмешательствах.
-
Дефицит АКТГ.
При нарушении функции гипоталамуса или гипофиза может возникать дефицит АКТГ, сопровождающийся обычно дефицитом и других гипофизарных гормонов в частности ГР и ТТГ. К дефициту АКТГ чаще всего приводят деструктивные процессы в области гипофиза, связанные, например, с краниофарингиомой и герминомой. Во многих случаях хирургическое удаление или лучевая терапия опухолей располагающихся по средней линии мозга, сопровождаются еще большим повреждением гипофиза. В очень редких случаях причиной дефицита АКТГ является аутоиммунный гипофизит.
Встречаются и врожденные дефекты гипофиза, которые метут распространяться на оседние структуры средней линии мозга, например зрительные нервы или прозрачную перегородку. В последнем случае патология носит название септооптической дисплазии, или синдрома де Морсъе. Гипофизарные расстройства могут быть следствием и более тяжелых нарушений развития головного мозга, таких как анэнцефалия и голопрозэнцефалия. Эти нарушения, как правило, носят спорадический характер, хотя наблюдались отдельные случаи их аутосомно- рецессивного наследования. В ряде семей у родных братьев и сестер был обнаружен изолированный дефицит АКТГ. Развитие прогрессирующей недостаточности АКТГ и кортизола бывает и при множественном дефиците гипофизарных гормонов вследствие мутаций гена PROP1. В арабских семьях встречается аутосомно-рецессивная изолированная недостаточность КРГ.
Клинические проявления
Поскольку при вторичной надпочечниковой недостаточности дефект кроется не в надпочечниках и ренин-ангиотензиновая система остается интактной, то секреция альдостерона сохраняется. Таким образом, признаки и симптомы этого состояния ограничиваются проявлениями дефицита кортизола — гипогликемией у новорожденных или ортостатической артериальной гипотонией и слабостью у детей более позднего возраста. Электролитный обмен, как правило, не нарушен.
У некоторых детей с патологией гипофиза наблюдаются лицевые аномалии по средней линии. При гипоплазии зрительных нервов имеются явные нарушения зрения, характерный блуждающий нистагм обычно обнаруживается лишь через несколько месяцев после рождения.
Лечение
Для предотвоащения ятрогенной вторичной надпочечниковой недостаточности (т. е. связанной с хроническим применением глюкокортикоидов) следует использовать наименьшие эффективные дозы синтетических гормонов и в течение как можно более короткого времени. Этого осложнения глюкокортикоидной терапии можно избежать быстрым снижением доз до уровня, эквивалентного физиологическим потребностям (примерно 10 мг/м2 в сутки), с последующим уменьшением их в течение нескольких недель. Такой режим обеспечивает восстановление функции коры надпочечников. Больные с анатомическими повреждениями гипофиза должны пожизненно получать глюкокортикоиды. Минералокортикоидная терапия им не нужна.

Врожденная гиперплазия коры надпочечников и родственные заболевания
Врожденная гиперплазия коры надпочечников (ВГКН) — это группа аутосомно-рецессивных наследственных нарушений биосинтеза кортизола. При дефиците кортизола возрастает секреция АКТГ, что приводит к гиперплазии коркового вещества надпочечников и, как следствие, усиленному образованию промежуточных продуктов стероидогенеза. В зависимости от нарушений конкретных ферментативных этапов этого процесса могут возникать признаки и симптомы дефицита или избытка минералокортикоидов, неполной вирилизации или преждевременного полового развития у мальчиков и вирилизации или полового инфантилизма у девочек.
Врожденная гиперплазия коры надпочечников вследствие недостаточности 21 -гидроксилазы
-
Генетика.
21-гидроксилаза стероидов кодируется двумя генами:
-
CYP21P (CYP21A1P, CYP21A);
-
CYP21 (CYP21A2, CYP21B);
расположенными на участке между локусами HLA-B и HLA-DR хромосомы 6р21.3 рядом с двумя генами 4-го компонента комплемента (JC4A и С4В). В этот кластер входят и многие другие гены. Активным геном 21-гидроксилазы является CYP21. Последовательность нуклеотидов в гене CYP21P на 98 % совпадает с последовательностью CYP21, но из-за 9 присутствующих в нем мутаций он не транскрибируется (псевдоген). Более 90 % мутаций, приводящих к недостаточности 21-гидроксилазы, представляют собой рекомбинации между CYP21 и CYP21P.
Повреждающие мутации псевдогена CYP21P, перенесенные на ген CYP21, по-разному сказываются на ферментативной активности. При некоторых из них синтез фермента вообще не происходит, другие же представляют собой миссенс-мутации, т. е. приводят к заменам аминокислот в структуре белка, при которых фермент сохраняет 1-50 % нормальной активности. Тяжесть заболевания зависит от характера мутаций. Так, больные с синдромом потери соли являются носителями мутаций в обоих аллелях гена, что полностью лишает белок ферментативной активности. У смешанных гетерозигот по разным мутациям тяжесть заболевания определяется в основном степенью сохранности менее поврежденного аллеля.
-
Клинические проявления.
Дефицит альдостерона и кортизола. Поскольку 21-гидроксилирование необходимо для синтеза, как кортизола, так и альдостерона, при самой тяжелой форме болезни, характеризующейся потерей соли, имеется дефицит обоих гормонов. На долю этой формы приходится около 75% случаев классической недостаточности 21-гидроксилазы. Проявления дефицита кортизола и альдостерона, как и механизмы развития соответствующих признаков и симптомов, в принципе не отличаются от стандартных. Они включают:
-
прогрессирующую потерю массы тела;
-
анорексию;
-
рвоту;
-
обезвоживание;
-
слабость;
-
артериальную гипотонию;
-
гипогликемию;
-
гипонатриемию;
-
гиперкалиемию.
Все эти симптомы обычно проявляются в 2-недельном возрасте. В отсутствие лечения уже через несколько дней или недель развиваются сосудистый коллапс, сердечные аритмии и наступает смерть.
От первичной надпочечниковой недостаточности, имеющей другие причины, ВГКН отличается тем, что при этом заболевании накапливаются предшественники стероидных гормонов, образующиеся на этапах биосинтеза, предшествующих заблокированной ферментативной реакции. Из-за дефицита кортизола возрастает секреция АКТГ, что приводит к гиперплазии коры надпочечников и увеличению уровня предшественников в сотни раз. При недостаточности 21-гидроксилазы возрастают концентрации 17-гидроксипрогестерона и прогестерона. Прогестерон и, вероятно, другие метаболиты действуют как антагонисты минералокортикоидных рецепторов и поэтому усиливают проявления дефицита альдостерона.
-
Избыток андрогенов в пренатальном периоде.
Накапливающийся в избытке 17-гидроксипрогестерон переходит на путь синтеза андрогенов. Это обусловливает секрецию надпочечниками большого количества андростендиона, который на периферии превращается в тестостерон. Усиленный синтез андрогенов при недостаточности 21-гидроксилазы начинается у плода на 8-10-й неделе внутриутробного периода, нарушая формирование женских наружных половых органов.
На ранних стадиях внутриутробного развития наружные половые органы плодов мужского и женского пола одинаковы. В норме у плода женского пола из полового бугорка образуется клитор, из уретральных складок — малые половые губы, а из губно-мошоночных складок — большие половые губы. У плода мужского пола наружные половые органы формируются под влиянием тестостерона, который секретируют яички. Половой бугорок увеличивается и образует головку полового члена, уретральные складки соединяются в тело полового члена и мочеиспускательный канал, а губно-мошоночные складки сращиваются, образуя мошонку. Под влиянием тестостерона происходит также превращение вольфовых протоков в мужские внутренние половые структуры — предстательную железу, семявыводящие пути и придатки яичек, но для этого необходим более высокий уровень тестостерона, чем для формирования наружных половых органов. На развитие женских внутренних половых структур (шейки и тела матки и маточных труб) тестостерон не влияет. У плодов мужского пола эти структуры атрофируются под действием фактора регрессии мюллеровых протоков, который секретируется яичками.
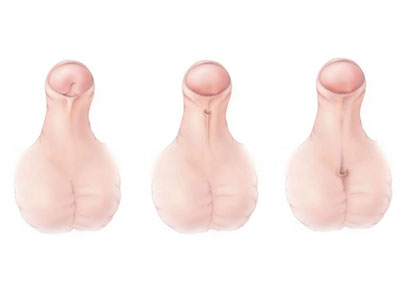
Таким образом, при секреции надпочечниками плода большого количества андрогенов у девочек происходит маскулинизация наружных половых органов. Это проявляется увеличением клитора и частичным или полным сращением половых губ. Влагалище и уретра обычно имеют общее отверстие (мочеполовой синус). Клитор может быть настолько велик, что напоминает половой член, а поскольку уретра открывается ниже, новорожденных девочек иногда принимают за мальчиков с гипоспадией и крипторхизмом. При недостаточности 21-гидроксилазы с потерей соли степень вирилизации еще больше. Внутренние половые органы остаются нормальными, так как у девочек из-за отсутствия яичек фактор peгрессии мюллеровых проток не вырабатывается.
У девочек, головной мозг которых во внутриутробном периоде подвергался воздействию высокого уровня андрогенов, может меняться половое поведение. Они часто играют с машинками, а не с куклами. В зрелом возрасте у них слабо выражен материнский инстинкт. Среди них много лесбиянок, но большинство все же гетеросексуальны и мало кто причисляет себя к мужчинам.
У мальчиков с этим заболеванием наружные половые органы имеют нормальное строение, и диагноз поэтому нередко устанавливают лишь после появления признаков надпочечниковой недостаточности. Поскольку болезнь протекает быстро, вероятность смерти у мальчиков выше, чем у девочек. В настоящее время во многих странах существуют программы массового обследования новорожденных на ВГКН.
-
Избыток андрогенов в постнатальном периоде.
В отсутствие правильного лечения у больных детей обоего пола развиваются дополнительные признаки избытка андрогенов. У мальчиков с простой вирилизирующей формой недостаточности 21-гидроксилазы болезнь обычно распознается поздно, поскольку при рождении они выглядят нормальными, а надпочечниковая недостаточность у них развивается редко.
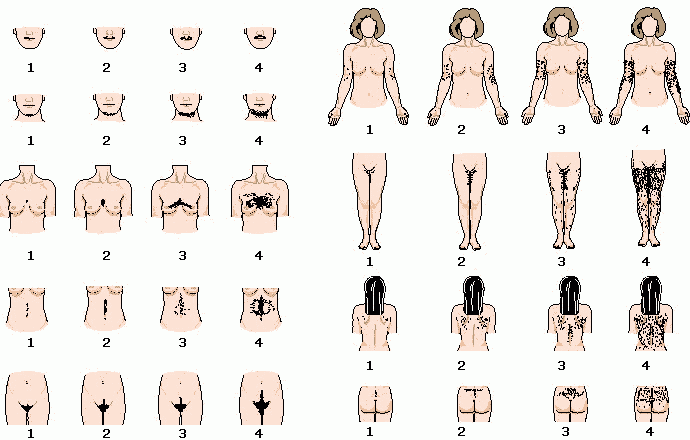
Признаки избыточной секреции андрогенов включают ускорение соматического роста и созревания костей. Поэтому в детском возрасте больные отличаются высоким ростом, но из-за преждевременного закрытия эпифизарных зон для больных зрелого возраста характерна низкорослость. Мышцы могут быть чрезмерно развиты, отмечается раннее лобковое и подмышечное оволосение, появляются угри и снижается тембр голоса. У мальчиков увеличиваются половой член, мошонка и предстательная железа, но яички обычно остаются маленькими, особенно в сравнении с размером полового члена. Иногда в яичках присутствуют эктопированные клетки коры надпочечника, которые гиперплазируются подобно клеткам самой коры и формируют опухоли яичек. У девочек продолжает увеличиваться клитор. Несмотря на сохранность внутренних половых органов, молочные железы не развиваются и менструации отсутствуют.
Сходные, но менее выраженные признаки избыточной секреции андрогенов наблюдаются и при неклассической форме недостаточности 21-гидроксилазы. В этих случаях уровень кортизола и альдостерона нормальный, наружные половые органы у девочек также имеют обычное строение. У детей обоего пола может иметь место преждевременное пубархе и раннее лобковое и подмышечное оволосение, а у женщин в дальнейшем — гирсутизм угревая сыпь, нарушение менструального цикла и бесплодие. Однако у многих больных заболевание протекает бессимптомно.
Лабораторные исследования
При синдроме потери соли все лабораторные показатели типичны для дефицита кортизола и альдостерона:
-
гипонатриемия;
-
гиперкалиемия;
-
ацидоз;
-
гипогликемия.
Однако эти сдвиги развиваются не ранее, чем через 1-2 недели после рождения. Содержание 17-гидроксипрогестерона в плазме значительно повышено, но в первые 2 -3 дня жизни оно высокое и у детей без недостаточности 21-гидроксилазы, особенно у больных или недоношенных. Суточный ритм 17-гидроксипрогестерона устанавливается одновременно с суточным ритмом кортизола и повторяет его. Уровень этого предшественника максимален по утрам и минимален ночью. Уровень кортизола в плазме у больных с синдромом потери соли, как правило, снижен. При простой вирилизирующей форме заболевания он часто остается нормальным, но оказывается явно ниже того, который должен быть при данном содержании АКТГ и 17-гидроксипрогестерона. Кроме 17-гидроксипрогестерона, в плазме больных девочек повышен уровень андростендиона и тестостерона. У мальчиков повышение уровня тестостерона выявить невозможно, поскольку у них в грудном возрасте содержание гормона и без того значительно выше, чем у детей более позднего возраста. Содержание 17-кетостероидов и прегнантриола в моче повышено, но в настоящее время эти показатели определяют редко, поскольку взять пробу крови у грудных детей гораздо легче, чем собирать суточную мочу. Уровень АКТГ повышен, но диагностическая ценность его определения не превышает ценности определения 17-гидроксипрогестерона. Активность ренина плазмы повышена, а содержание альдостерона в крови оказывается слишком низким, не соответствующим активности ренина. Однако в первые дни жизни активность ренина высока и у здоровых детей.
Наиболее надежным диагностическим методом для выявления недостаточности 21-гидроксилазы является определение уровня 17-гидроксипрогестерона до и через 30 или 60 мин после одномоментного внутривенного введения 0,125-0,25 мг тетракозактина. Существуют номограммы, по которым здоровых детей легко отличить от больных с неклассической и классической недостаточностью 21-гидроксилазы. У гетерозиготных носителей этого аутосомно-рецессивного заболевания содержание 17-гидроксипрогестерона после введения АКТГ обычно возрастает несколько больше, чем у детей без генетического дефекта, хотя показатели в значительной степени перекрываются.
-
Дополнительные исследования.
У ребенка с наружными половыми органами промежуточного типа, прежде всего, необходимо тщательно исследовать их строение, найти отверстие мочеиспускательного канала, прощупать мошонку или половые губы и паховые области, а также выяснить наличие других анатомических аномалий. Если удается нащупать гонады, то это почти всегда яички, т. е. генетический пол ребенка — мужской. Присутствие или отсутствие матки и локализацию гонад устанавливают с помощью УЗИ. Для быстрого установления генетического пола ребенка определяют кариотип, исследуя X- и Y-хромосомы в интерфазных ядрах методом FISH. Все эти данные позволяют сообщить родителям генетический пол и особенности строения наружных половых органов ребенка еще до получения результатов гормональных исследований. При женском псевдогермафродитизме обнаружение матки и влагалища путем введения контраста в мочеполовой синус помогает выработке плана хирургического лечения.
Врожденная гиперплазия коры надпочечников вследствие недостаточности
-
Этиология.
В основе недостаточности 11р-гидроксилазы лежит мутация гена CYP11B1, расположенного на хромосоме 8q24. Этот ген кодирует фермент, катализирующий гидроксилирование 11-го углеродного атома 11-дезоксикортизола, приводящее к образованию кортизола. При недостаточности фермента кортизол не образуется и возрастает уровень АКТГ. Вследствие этого накапливаются предшественники, особенно 11-дезоксикортизол и дезоксикортикостерон, которые вступают на путь синтеза андрогенов, как это происходит и при недостаточности 21-гидроксилазы. Однако в этих случаях ген CYP11B2, кодирующий альдостеронсинтазу, не поврежден и синтез альдостерона не нарушается.
Недостаточность 11р-гидроксилазы отвечает за 5-8% случаев ВГКН и встречается с частотой 1:100 000-250 000 новорожденных. Выявлено более 30 различных мутаций гена CYP11B1. Болезнь наиболее распространена среди израильских евреев, выходцев из Северной Африки (1:15 000-17 000). Почти все больные этой этнической группы являются носителями мутации, приводящей к замене аргинина в положении 448 на гистидин (R448H) в молекуле фермента. Недостаточность 11р-гидроксилазы проявляется тяжелой классической и, реже, более легкой неклассической формой.
-
Клинические проявления.
Хотя образование кортизола резко снижается, синтез альдостерона сохраняется. При интактности альдостеронсинтазы из прогестерона образуется и некоторое количество кортикостерона. Поэтому у больных практически отсутствуют такие признаки надпочечниковой недостаточности, как артериальная гипотония, гипогликемия, гипонатриемия и гиперкалиемия. Примерно у 60-70% больных развивается артериальная гипертония, но они в течение многих лет могут обходиться без лечения. Артериальная гипертония обусловливается, по-видимому, высоким уровнем дезоксикортикостерона, который обладает минералокортикоидной активностью, или его метаболитов. После начала лечения гидрокортизоном могут временно возникать признаки минералокортикоидной недостаточности. Причиной этого служит, вероятно, острое подавление секреции дезоксикортикостерона на фоне атрофии клубочковой зоны в условиях хронического снижения активности ренина плазмы.
Для недостаточности 11р-гидроксилазы характерны те же признаки и симптомы, что и для 21 -гидроксилазы.
-
Лабораторные исследования.
В плазме повышен уровень 11-дезоксикортизола и дезоксикортикостерона. Поскольку дезоксикортикостерон и его метаболиты обладают минералокортикоидной активностью, активность ренина плазмы падает, что приводит к снижению и уровня альдостерона, несмотря на сохранность ферментов его синтеза. Иногда развивается гипокалиеиический алкалоз.
-
Лечение.
Применяют гидрокортизон в тех же дозах, что и при недостаточности 21-гидроксилазы. У больных грудного возраста иногда возникает необходимость в кратковременном применении минералокортикоидов. В остальных случаях это обычно не требуется. Артериальная гипертония часто устраняется приемом глюкокортикоидов, но при длительной гипертонии приходится прибегать и к другим средствам, лучше всего использовать блокаторы кальциевых каналов.
Врожденная гиперплазия коры надпочечников вследствие недостаточности 3Р-гидроксистероиддегидрогеназы
-
Этиология.
Недостаточность ЗР-гидроксистероиддегидрогеназы встречается менее чем у 2 % больных с ВГКН. Этот фермент необходим для превращения А5-стероидов (прегненолона, 17-гидроксипрегненолона, ДЭА) в А4-стероиды (прогестерон, 17-гидроксипрогестерон и андростендион). При недостаточности фермента нарушается синтез кортизола, альдостерона и андростендиона, но возрастает секреция ДЭА. ЗР-гидроксистероиддегидрогеназа экспрессируется в коре надпочечников и половых железах и кодируется геном HSD3B2, расположенным на хромосоме 1. У больных с недостаточностью этого фермента обнаружено более 30 различных мутаций его гена.
-
Клинические проявления.
При классической форме болезни не синтезируется ни кортизол, ни альдостерон, поэтому у больных грудного возраста могут возникать кризы, связанные с потерей соли. Дефицит андростендиона и тестостерона приводит к недостаточной вирилизации мальчиков. У них наблюдаются гипоспадии, а иногда и раздвоение мошонки и крипторхизм. Из-за повышения уровня ДЭА, обладающего слабой андрогенной активностью, у девочек отмечается некоторая вирилизация с увеличением клитора. Избыточная секреция ДЭА может быть причиной преждевременного адренархе. В подростковом и зрелом возрасте у женщин наблюдаются нарушения менструального цикла и синдром поликистоза яичников. Для мальчиков характерна та или иная степень гипогонадизма, хотя появление вторичных половых признаков не исключено. О дефекте фермента в яичках свидетельствует высокое отношение А5:А4-стероидов в сперме.
-
Лабораторные исследования.
Основной признак данного заболевания — значительное повышение в плазме уровня А5-стероидов (17-гидроксипрегненолона и ДЭА), образующихся выше заблокированной ферментативной реакции. Поскольку вне надпочечников и гонад активность ЗР-гидроксистероиддегидрогеназы сохраняется, у больных может возрастать также уровень17-гидроксипрогестерона, что иногда приводит к ошибочному диагнозу недостаточности 21-гидроксилазы. При заболевании, сопровождающемся потерей соли, повышена активность ренина плазмы.
Легкое или умеренное повышение уровня ДЭА в сыворотке крови часто обнаруживается у детей с преждевременным адренархе и у женщин с признаками избыточной секреции андрогенов. Считается, что при этом имеется неклассическая форма недостаточности ЗР-гидрохсистероиддегидрогеназы. Однако в большинстве таких случаев не находят мутаций гена HSD3B2. По-видимому, истинная неклассическая форма этой болезни встречается крайне редко.
-
Лечение.
Как и при недостаточности 21-гидроксилазы, больные нуждаются в заместительной глюкокортикоидной и минералокортикоидной терапии соответственно гидрокортизоном и флудрокортизоном. При неполной вирилизации у больных, которых собираются воспитывать как мальчиков, размеры полового члена можно увеличить несколькими инъекциями депо-препаратов тестостерона в раннем грудном возрасте. Введение тестостерона может оказаться необходимым и в период полового развития.
Врожденная гиперплазия коры надпочечников вследствие недостаточности 17-гидроксилазы
-
Этиология.
Недостаточность 17-гидроксилазы лежит в основе менее 1 % случаев ВГКН. Один и тот же фермент CYP17 катализирует две разных. Врожденная гиперплазия коры надпочечников и родственные заболевания реакции:
-
во-первых, 17-гидроксилирование прегненолона и прогестерона с образованием соответственно 17-гидроксипрегненолона и 17-гидроксипрогестерона;
-
во-вторых, 17,20-лиазную реакцию, в результате которой 17-гидроксипрегненолон превращается в ДЭА и (в меньшей степени) 17-гидроксипрогестерон превращается в А4-андростендион.
ДЭА и андростендион служат предшественниками тестостерона и эстрогенов. 17-гидроксилаза экспрессируется в коре надпочечников и половых железах и кодируется геном, расположенным на хромосоме 10. Большинство мутаций этого гена сказывается, как на гидроксилазной, так и на лиазной активности фермента, но некоторые снижают только одну из его активностей.
-
Клинические проявления и лабораторные исследования.
У больных с недостаточностью 17-гидроксилазы не синтезируется кортизол, но сохраняется способность к синтезу кортикостерона. Поскольку кортикостерон обладает относительно высокой глюкокортикоидной активностью, надпочечниковая недостаточность в таких случаях не развивается. Однако в избытке образуется и непосредственный предшественник кортикостерона — дезоксикортикостерон, что может привести к развитию артериальной гипертонии, гипокалиемии и снижению секреции ренина и альдостерона, как при недостаточности 11-гидроксилазы. В то же время, в отличие от последнего заболевания, у больных с недостаточностью 17-гидроксилазы снижается синтез половых гормонов. Мальчики в этих случаях имеют женский фенотип (но в половых губах или паховых областях прощупываются гонады) или наружные половые органы промежуточного типа (мужской псевдогермафродитизм). У девочек обычно пубертатный период не наступает в соответствующее время. Возможность недостаточности 17-гидроксилазы у девочек следует учитывать при дифференциальной диагностике первичного гипогонадизма. Помимо повышения уровня дезоксикортикостерона, снижения активности ренина и содержания альдостерона и 17-гидроксилированных стероидов, для недостаточности 17-гидроксилазы характерно отсутствие реакции 1 кортизола и половых стероидов на введение АКТГ и ХГ соответственно.

-
Лечение.
Для подавления секреции дезоксикортикостерона и, тем самым, устранения артериальной гипертонии при недостаточности 17-гидроксилазы необходима заместительная глюкокортикоидная терапия. Могут потребоваться и другие гипотензивные средства. Девочкам пубертатного возраста нужна заместительная терапия эстрогенами. Детям генетически мужского пола в зависимости от выбранного пола воспитания вводят либо андрогены, либо эстрогены. Как и при синдроме резистентности к андрогенам, у генетических мальчиков, которых собираются воспитывать как девочек, во время или до начала полового созревания необходима гонадэктомия во избежание злокачественного перерождения яичек, находящихся в брюшной полости.








- Аллергия
- Ангиология
- Болезни глаз
- Венерология
- Гастроэнтерология
- Гинекология
- Дерматология
- Здоровое питание
- Инфекционные болезни
- Кардиология
- Косметология
- Лекарства
- Лекарственные растения
- ЛОР-заболевания
- Мужское здоровье
- Неврология
- Неотложная помощь
- Новости
- Онкология
- Ортопедия
- Паразитология
- Педиатрия
- Пульмонология
- Расшифровка анализов
- Симптомы
- Системные заболевания
- Стоматология
- Травматология
- Урология
- Хирургия
- Эндокринология
- Нужно знать
- Еда
- Профессиональные заболевания
Комментарии