
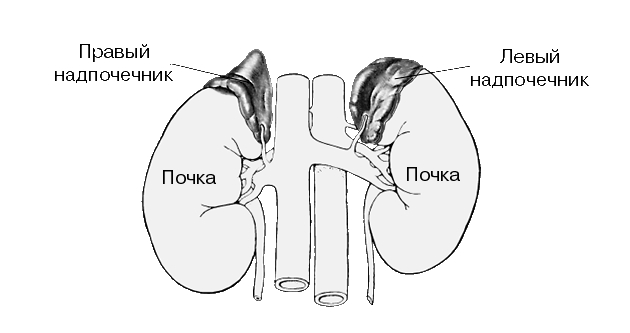
Первичная надпочечниковая недостаточность у детей: причины, симптомы, лечение
Первичная надпочечниковая недостаточность может иметь генетические причины и, хотя не обязательно, проявляется в грудном возрасте. Она может быть и приобретенной, например, при аутоиммунном поражении коры надпочечников. Однако предрасположенность к аутоиммунной патологии часто также имеет генетическую природу, и поэтому такое разграничение весьма относительно.
-
Врожденная надпочечниковая недостаточность.
Врожденные дефекты стероидогенеза. Самая частая причина надпочечниковой недостаточности в грудном возрасте — врожденная гиперплазия коры надпочечников с потерей соли. Примерно у 75% детей с недостаточностью 21-гидроксилазы, почти у всех детей с липоидной гиперплазией коры надпочечников и у большинства детей с недостаточностью ЗР-гидроксистероиддегидрогеназы после рождения проявляются симптомы потери соли, поскольку в этих случаях нарушен синтез не только кортизола, но и альдостерона.
-
Врожденная гипоплазия надпочечников.
Надпочечниковая недостаточность в таких случаях обычно проявляется острыми симптомами сразу после рождения, но иногда они возникают лишь в позднем детстве или даже в зрелом возрасте. При гистологическом исследовании гипоплазированного коркового вещества надпочечников обнаруживается его дезорганизация и цитомегалия. Эта патология встречается в основном у мальчиков и обусловлена мутацией гена DAX1, расположенного на хромосоме Хр21 и участвующего в синтезе ядерных гормональных рецепторов. У мальчиков с таким дефектом из-за вторичного гипогонадизма отсутствует половое развитие. В основе вторичного гипогонадизма лежит мутация того же гена DAX1. Крипторхизм, который часто наблюдается у таких мальчиков, является, вероятно, ранним признаком вторичного гипогонадизма.
Врожденная гипоплазия надпочечников может быть также проявлением синдрома генных последовательностей, сочетаясь с мышечной дистрофией Дюшенна, недостаточностью глицерокиназы и/или умственной отсталостью.
-
Адренолейкодистрофия.
При этой патологии надпочечниковой недостаточности сопутствует демиелинизация нейронов ЦНС. Из-за нарушения пероксисомного окисления в тканях и биологических жидкостях накапливаются жирные кислоты с очень длинной цепью.
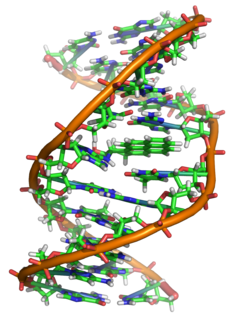
Наиболее распространена Х-сцепленная форма адренолейкодистрофии (Х-АЛД) с различной пенетрантностью. Болезнь чаще всего проявляется дегенеративными неврологическими расстройствами в детском или подростковом возрасте, прогрессирующими до тяжелой деменции с нарушением зрения, слуха, речи и походки. Через несколько лет наступает смерть. Более легкая форма Х-АЛД — адреномиелоневропатия — начинается в позднем подростковом или раннем зрелом возрасте. Надпочечниковая недостаточность часто развивается задолго до появления неврологических симптомов и может быть единственным проявлением болезни. В основе этого заболевания лежат мутации гена ABCD1, расположенного на хромосоме Xq28. Ген кодирует трансмембранный транспортер, участвующий в переносе жирных кислот с очень длинной цепью в пероксисомы.
У больных с Х-АЛД обнаружено более 400 мутаций этого гена. У членов одной и той же семьи обычно выявляется одна и та же мутация, хотя болезнь может протекать по-разному. Это связано, вероятно, с действием генов-модификаторов или какими-то иными факторами. Степень неврологических расстройств и тяжесть надпочечниковой недостаточности не соответствуют друг другу. В настоящее время возможна пренатальная диагностика этого заболевания с помощью анализа ДНК или определения уровня жирных кислот с очень длинной цепью у родственников. У женщин — гетерозиготных носителей дефектного гена симптомы заболевания возникают относительно поздно. Надпочечниковая недостаточность развивается редко. Для лечения используют:
-
глицерин триолеат или глицерина триэрукат (масло Лоренцо);
-
пересадку костного мозга;
-
ловастатин;
-
фенофибрат;
-
генную терапию.
Однако эффективность всех этих средств и методов пока неизвестна.
АЛД новорожденных — редкое аутосомно-рецессивное заболевание. Оно характеризуется неврологическими расстройствами и нарушением функции коры надпочечников. У большинства больных наблюдается тяжелая умственная отсталость, и они погибают до 5-летнего возраста. Это заболевание представляет собой разновидность синдрома Зельвегера (цереброгепаторенального синдрома), при котором из-за мутаций тех или иных генов, контролирующих образование пероксисом, последние полностью отсутствуют.
-
Семейная недостаточность глюкокортикоидов.

Эта форма хронической надпочечниковой недостаточности характеризуется изолированным дефицитом глюкокортикоидов, повышенным уровнем АКТГ и нормальной секрецией альдостерона. Потеря соли, наблюдаемая при большинстве других форм надпочечниковой недостаточности, в данном случае отсутствует. В раннем возрасте у больных развивается гипогликемия и возникают судороги, отмечается усиленная пигментация кожи. У мальчиков и девочек это аутосомно-рецессивное заболевание встречается с равной частотой. Кора надпочечников атрофирована, но ее клубочковая зона относительно сохранена. У некоторых (около 40%), но не у всех больных обнаруживаются различные мутации гена, кодирующего рецептор АКТГ.
Резистентность к АКТГ наблюдается и при синдроме Оллгрова (синдроме трех А), при котором такая резистентность сочетается с ахалазией кардиального отдела желудка и алакримией. При этом заболевании часто имеют место нарушения функции автономной нервной системы, умственная отсталость, глухота и моторная невропатия. Синдром Оллгрова также наследуется аутосомно- рецессивным способом; ген картирован на хромосоме 12ql3.
-
Нарушения синтеза и метаболизма холестерина.
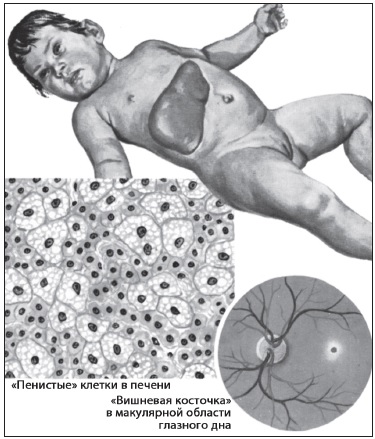
К заболеваниям этой группы относится абеталипопротеидемия с недостаточностью апоВ-содержащих липопротеидов, и семейная гиперхолестеринемия с нарушением рецепторов ЛПНП У больных с такой патологией обнаружено снижение функции коры надпочечников. Надпочечниковую недостаточность находили при синдроме Смита-Лемли -Опица, аутосомно-рецессивном заболевании с лицевыми аномалиями, микроцефалией, аномалиями конечностей и задержкой развития. При этом синдроме выявлены мутации расположенного на хромосоме Ilql2-ql3 гена, который кодирует Д7-редуктазу стеролов. В результате нарушается конечный этап синтеза холестерина и снижается его уровень, резко возрастает содержание 7-дегидрохолестерина и развивается надпочечниковая недостаточность. Болезнь Вольмана – редкая аутосомно-рецессивная патология, в основе которой лежат мутации гена, кодирующего кислую липазу лизосом. Это приводит к накоплению эфиров холестерина в лизосомах большинства органов и, в конце концов, к нарушению их функции. У детей на 1-2-м месяце жизни отмечается гепатоспленомегалия, стеаторея, вздутие живота и отставание в развитии. Выявляется надпочечниковая недостаточность и двусторонняя кальцификация надпочечников. Больные обычно погибают на первом году жизни. Ген, кодирующий кислую липазу лизосом, расположен на хромосоме 10q23.2-23.3, и его мутации при болезни Вольмана известны.
-
Недостаточность транскортина и снижение его сродства к кортизолу.
При этой патологии уровень кортизола в плазме снижен, но содержание свободного кортизола в моче и уровень АКТГ в плазме нормальные. У взрослых с недостаточностью транскортина часто отмечается артериальная гипотония и повышенная утомляемость.
Приобретенная надпочечниковая недостаточность
-
Аутоиммунная болезнь Аддисона.
Приобретенная первичная надпочечниковая недостаточность чаще всего связана с аутоиммунной деструкцией желез. Надпочечники подчас настолько уменьшаются в размерах, что не видны при аутопсии, а на микроскопических срезах удается обнаружить лишь остатки их ткани. Мозговое вещество, как правило, сохраняется, а на месте коркового вещества в изобилии присутствуют лимфоциты. В запущенных случаях выпадает секреция всех гормонов коркового вещества, но на ранних стадиях возможна только недостаточность кортизола. У большинства больных в плазме присутствуют антитела к цитоплазматическим антигенам клеток коркового вещества, аутоантигеном чаще всего является 21-гидроксилаза (CYP21).
Болезнь Аддисона нередко представляет собой компонент двух аутоиммунных полигландулярных синдромов. Первым проявлением аутоиммунного полигландулярного синдрома типа I обычно бывает хронический кандидоз кожи и слизистых оболочек. Затем развиваются гипопаратиреоз и, наконец, первичная надпочечниковая недостаточность, которая в типичных случаях проявляется в раннем подростковом возрасте. К другим обычно сопутствующим аутоиммунным нарушениям относятся недостаточность половых желез, алопеция, витилиго, кератопатия, гипоплазия зубной эмали, дистрофия ногтей, нарушение процессов всасывания в кишечнике и хронический активный гепатит. Гипотиреоз и сахарный диабет типа I встречаются менее чем у 10% больных. Некоторые компоненты этого синдрома могут проявляться даже после 40 лет. Наличие у больных антител к ткани надпочечников и к стероидпродуцирующим клеткам указывает на высокую вероятность развития болезни Аддисона или (у женщин) недостаточности яичников. Надпочечниковая недостаточность при аутоиммунном полигландулярном синдроме типа I может развиваться достаточно быстро. Описаны случаи неожиданной смерти больных и их братьев или сестер, что подчеркивает необходимость тщательного наблюдения за больными и детального обследования даже их внешне здоровых ближайших родственников.
Среди аутоантител к ферментам стероидогенеза обнаруживаются антитела к CYP21, CYP17 HCYPHAI. Синдром наследуется аутосомно-рецессивно, а дефект выявлен в расположенном на хромосоме 21q22.3 гене, получившем название аутоиммунного регулятора-1 (AIRE1). Этот ген кодирует фактор транскрипции, играющий важную роль в регуляции иммунного ответа. У больных с аутоиммунным полигландулярным синдромом типа I установлено примерно 40 разных мутаций гена AIRE1, причем две из них (R257X и деления трех пар оснований) встречаются наиболее часто. В одной семье с миссенсмутацией G228W заболевание наследовалось аутосомно-доминантным путем.
Аутоиммунный полигландулярный синдром типа II представляет собой сочетание хронической надпочечниковой недостаточности с аутоиммунным поражением щитовидной железы (синдром Шмидта) или сахарным диабетом типа I (синдром Карпентера). Этим нарушениям иногда сопутствуют недостаточность половых желез, витилиго, алопеция, а также хронический атрофический гастрит с болезнью Аддисона-Бирмера или без нее. Среди таких больных с повышенной частотой встречаются HLA-D3 и HLA-D4 (маркеры риска). С аутоиммунным полигландулярным синдромом типа II ассоциированы также гены МНС класса I MICA и MICB. Этот синдром характерен в основном для женщин среднего возраста и может передаваться из поколения в поколение. У больных также обнаруживаются антитела к надпочечниковым антигенам CYP21, CYP17 и CYP11A1.
-
Инфекции.
В прошлом деструкция надпочечников очень часто была связана с их туберкулезным поражением, но в настоящее время это встречается гораздо реже. Наиболее распространенная инфекционная причина надпочечниковой недостаточности — менингококкемия. Гипоадреналовый криз, развивающийся при молниеносном менингококковом сепсисе, называют синдромом Уотерхауса-Фридериксена. У больных СПИДом могут иметь место различные нарушения ГГНС, но явная надпочечниковая недостаточность развивается редко. Однако лекарственные средства, применяемые при СПИДе, могут влиять на функцию надпочечников.
-
Лекарственные средства.
Противогрибковое средство кетоконазол вызывает надпочечниковую недостаточность, ингибируя активность ферментов стероидогенеза. Рифампицин и противосудорожные препараты (фенотоин и фенобарбитал) уменьшают эффективность заместительной кортикостероидной терапии, индуцируя в печени синтез ферментов стероидного метаболизма. Митотан, применяемый при раке надпочечников и синдроме Кушинга, оказывает токсическое действие на корковое вещество надпочечников, а также влияет на периферический метаболизм кортизола. Признаки надпочечниковой недостаточности появляются у значительного числа больных, получающих митотан.
-
Кровоизлияние в надпочечники.
У новорожденных это может быть следствием трудных родов (особенно при ягодичном предлежании плода), но установить причину не всегда удается. Такие случаи встречаются с частотой 3:100 000 родившихся живыми детей. Иногда кровоизлияние бывает настолько значительным, что приводит к смерти от кровопотери или острой надпочечниковой недостаточности. При кровоизлиянии в надпочечники прощупывается образование в животе, развивается необъяснимая желтуха или гематома мошонки. Нередко симптомы появляются лишь после кальцификации надпочечников. Описаны случаи кровоизлияния в надпочечники еще во внутриутробном периоде. Самая частая причина кровоизлияния в постнатальной жизни — прием антикоагулянтов. У детей это может быть следствием случайного отравления такими средствами.








- Аллергия
- Ангиология
- Болезни глаз
- Венерология
- Гастроэнтерология
- Гинекология
- Дерматология
- Здоровое питание
- Инфекционные болезни
- Кардиология
- Косметология
- Лекарства
- Лекарственные растения
- ЛОР-заболевания
- Мужское здоровье
- Неврология
- Неотложная помощь
- Новости
- Онкология
- Ортопедия
- Паразитология
- Педиатрия
- Пульмонология
- Расшифровка анализов
- Симптомы
- Системные заболевания
- Стоматология
- Травматология
- Урология
- Хирургия
- Эндокринология
- Нужно знать
- Еда
- Профессиональные заболевания
Комментарии