
Морфология легочной гипертензии. Патофизиология порока
Гипоксия как фактор, вызывающий сужение легочных сосудов, известна более 100 лет. Точный механизм гипоксической вазоконстрикции неизвестен. Локальная гипоксия легких действует как гомеостатический механизм, направленный на отведение легочного кровотока от плохо вентилируемых участков легких, поддерживая вентиляционно-перфузионные соотношения. Этим объясняется гипоксическая легочная вазоконстрикция. В ответ на гипоксический эпизод давление в легочной артерии повышается в течение нескольких секунд. Эта реакция сосудов выражена при альвеолярной гипоксии более ярко, чем при легочно-артериальной гипоксии. Предполагается, что гипоксия приводит к деполяризации мембранного потенциала гладкомышечной клетки сосуда, стимулируя вхождение кальция в клетку через потенциалзависимые кальциевые каналы. Ни один из известных вазоактивных медиаторов – катехоламины, гистамин, серотонин, ангиотензин II, брадикинин, простагландины, тромбоксан, вазопрессин, ацетилхолин, вазоактивный кишечный пептид, субстанция Р, АТФ, тромбоцитоактивирующий фактор – не оказались причастными к гипоксической вазоконстрикции. Последующие исследования показали, что гипоксическая легочная вазоконстрикция частично обусловлена притоком кальция в клетки через потенциалзависимые каналы и инициирована гипоксическим угнетением выхода калия, который вызывает мембранную деполяризацию. Реакция сосудов на гипоксию различна и зависит от многих факторов, включая степень мускуляризации легочного сосудистого русла, базовый сосудистый тонус, пол, концентрацию водородных ионов, гематокрит.
Гипоксическая легочная вазоконстрикция
Хроническая гипоксия приводит к ремоделированию легочных сосудов, механизмы которого неизвестны. Оно проявляется гипертрофией мышечного слоя дистальных легочных артерий и, как следствие, – легочной гипертензией. Гипоксическая полицитемия также способствует повышению давления в легочных сосудах. Хроническая гипоксия способствует распространению гладкой мускулатуры дистально на легочные артериолы диаметром менее 100 мкм, которые в норме содержат только эластический слой. Кроме гипоксии изменения микрососудов вызывает сама легочная гипертензия. Легочный гипертонический синдром, связанный с гипоксией, в эксперименте на животных удавалось смягчить такими субстанциями, как гепарин, блокаторы кальциевых каналов, р-блокаторы, ингибиторы ангиотензин-превращающего фермента, ингибиторы циклооксигеназы и липок- сигеназы, мусорщики гидроксильных радикалов, ингибиторы орнитиновой декарбоксилазы и антагонисты эндотелина. Механизмы ингибирования пролиферации гладкой мускулатуры этими субстанциями различны, что свидетельствует о существовании множественных факторов, способствующих развитию гипоксической ЛГ.
Реакция на острую гипоксию быстрая и обратимая, в то время как ответ на хроническую гипоксию может быть частично необратимым и требует более продолжительного времени для возвращения к нормальному состоянию.
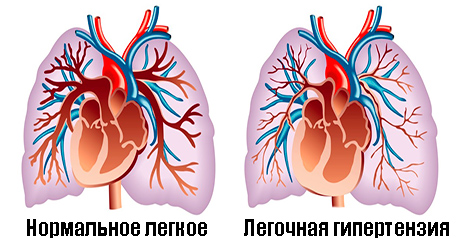
Гипоксия и связанное с ней увеличение клеточной пролиферации сопровождяется увеличением синтеза ДНК, продукции матриксных белков, усилением эластолитической активности и изменением фенотипа синтеза белков, отличных от сократительных белков актина и миозина, в пользу синтеза внеклеточных матриксных белков. Гипоксия может также стать причиной снижения количества ß-рецепторов и способности продуцировать цАМФ.
Структурная основа ЛГ
В 1958 г. Heath и Edwards дали классическое описание прогрессирующего характера структурных изменений легочных сосудов, разделив их на 6 стадий.
Стадии I и II являются начальными и, возможно, обратимыми. Стадия III характеризуется окклюзирующими просвет изменениями – дальнейшим утолщением медии за счет пучков продольных мышц, гиперплазии интимы и фиброзно-эластической ткани. Стадия IV – истончение медии, дилатация сосуда, окклюзия его просвета фиброзной тканью. Стадия III в лучшем случае частично обратимая, IV – абсолютно необратима. Стадии V и VI являются терминальными – возникают ангиоматозные изменения в виде множественных тонкостенных сосудов, которые продолжаются в капилляры стенок альвеол, фибриноидный некроз и тяжелый реактивный воспалительный эксудат во всех слоях сосуда. Терминальные стадии чаще обнаруживаются у детей старшего возраста.
Структурные изменения, которые соответствуют высокому и фиксированному ЛСС, реже обнаруживаются у младенцев и детей младшего возраста, хотя они отмечаются при некоторых ВПС, таких, как ТМА с ДМЖП и даже с интактной межжелудочковой перегородкой. Любопытно, что прогрессирование ОБЛС отмечали даже после успешной хирургической коррекции ТМА. У младенцев с большим ДМЖП обструктивная болезнь развивается рано. У детей с АВСД она особенно быстро прогрессирует даже в возрасте до 6 мес. Характерно, что раннее развитие неоперабельности отмечается при пороках, гемодинамической особенностью которых является сочетание гиперволемической гипертензии и гипоксемии. Кроме перечисленных пороков к ним относятся ОАС, единственный желудочек сердца и атрезия трехстворчастого клапана с гипертензией, ТАДЛВ. Скорость развития ОБЛС даже в пределах одной нозологической формы индивидуальна, несмотря на сходные гемодинамические условия. Причина этого неизвестна. Не до конца выяснены причины быстрого прогрессирования ОБЛС у детей с синдромом Дауна. Возможно, это связано с характерными для них частыми респираторными инфекциями и ночной обструкцией верхних дыхательных путей, усиливающими гипоксический компонент.
Риск возникновения ОБЛС существует также у больных с цианотическими пороками сердца при наличии хирургически созданных системно-легочных шунтов.
Ряд исследователей пытались дать количественную оценку степени гипертрофии медии, однако эти измерения не коррелировали тесно с дооперационным уровнем ЛСС и с его изменениями после операции. С возрастом структура ацинарных артерий изменяется за счет распространения мышечного слоя к периферии. Артерии, не содержавшие мышечного слоя, становятся частично мышечными, а позднее – полностью мускуляризированными. При рождении мышечные артерии толстостенны, однако в течение нескольких дней самые узкие из них расширяются, достигая такого же диаметра, как у взрослых. К 4-месячному возрасту этот процесс завершается вовлечением артерии большего диаметра. Увеличиваются количество и размеры артерий; особенно быстро это происходит в младенческом возрасте. Хотя альвеолы также пролиферируют, соотношение альвеол к артериям уменьшается с 20:1 у новорожденных до 8:1 в раннем детском возрасте, сохраняясь таковым в последующем.
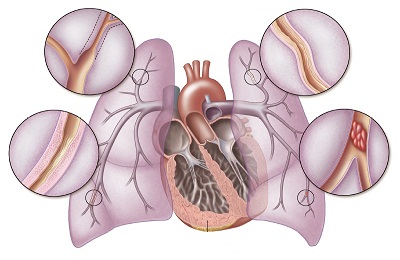
При ДМЖП с ЛГ на ангиограммах артерии в корне легких дилатированы и сужены к периферии. При микроскопических исследованиях легочного сосудистого русла гладкая мускулатура медиального слоя быстро распространяется на сосуды, которые в норме не являются мышечными. Перинатальная регрессия гипертрофии гладкой мускулатуры не происходит, и наоборот, гипертрофия медии увеличивается. Нарушается нормальный рост артериальной сети, в то же время дифференциация и увеличение числа альвеол происходят без отклонения от нормы. Морфометрический метод исследования характера мускуляризации, числа и размеров артериальных микрососудов в биоптатах легких имеет преимущества перед другими морфологическими исследованиями, так как изменения во всех отделах легких одинаковы.
Структура эндотелиальных клеток при ЛГ
При электронной микроскопии легочных биоптатов, взятых у пациентов, у которых диагностирован ВПС в сочетании с ЛГ, обнаружены структурные изменения эндотелиальных клеток. Они являются причиной дисфункции эндотелия, которая проявляется повышением реактивности легочных сосудов и играет важную роль в патогенезе прогрессирующей болезни легочных сосудов. При сканирующей электронной микроскопии у детей с увеличенным легочным кровотоком уже к 2-месячному возрасту поверхность эндотелия напоминает гладкомышечные клетки – эндотелиальные клетки вытягиваются в направлении кровотока.
Таким образом, уже на ранней стадии течения порока цитоскелет нарушен. Эндотелиальная поверхность гипертензивных толстостенных легочных артерий имеет канатовидную текстуру, когда клетки образуют скрученные гребни с глубокими впадинами. У пациентов с выраженными изменениями легочных сосудов эндотелиальная поверхность напоминает бахрому – высокие гребни чередуются с узкими глубокими скрученными щелями, поэтому эндотелий гипертензивных легочных сосудов представляет собой грубую выстилку по сравнению с эндотелием нормотензивных артерий и нарушает нормальное взаимодействие пристеночных элементов крови – тромбоцитов и лейкоцитов – с сосудистой стенкой. Это приводит к освобождению вазоконстрикторных субстанций и усилению митогенеза в гладкомышечных клетках. Субэндотелий мышечных артерий также изменен за счет деградации и неосинтеза внутренней эластической пластинки.
Патогенез структурных изменений легких при ВПС с гипертензией малого круга кровообращения
Выше был проведен подробный анализ физиологических механизмов регулирования легочного кровотока, а также событий, развивающихся в МКК при ЛГ. Важно проследить последовательность и взаимосвязь этих событий, представить основные виды структурных изменений легких при хронической перегрузке артериального и венозного звеньев МКК.
Прежде всего, следует подчеркнуть два очевидных, но часто упускаемых из вида основополагающих обстоятельства. Во-первых, главной функциональной единицей кровеносной системы является капилляр, так как именно на уровне капилляров и частично – посткапиллярных венул осуществляется обмен веществ между кровью и тканями в большом круге кровообращения и между кровью и воздухом альвеол в МКК. Поэтому все местные и системные регуляторные механизмы сердечно-сосудистой системы направлены прямо или опосредованно на поддержание в капиллярах оптимальной для метаболизма скорости кровотока. Замедление продвижения эритроцитов по капилляру привело бы в БКК к элиминации кислорода из крови в ткань на артериальном конце капилляра, тогда как дистальный сегмент оказался бы в условиях гипоксии, от чего пострадали бы не только тканевые элементы, но и сами капиллярные структуры, с целым каскадом последующих изменений. Ускоренное движение крови по капиллярам практически эквивалентно шунтирующему кровотоку, когда эритроциты и компоненты плазмы минуют зону микроциркуляторого русла, пригодную для метаболизма, не успев в полной мере реализовать этот метаболизм. Такая ситуации возникает при значительном повышении давления крови на артериальном конце капилляра. Она также чревата опасностью перерастяжения капилляров многорядным потоком эритроцитов, из которых элементы, находящиеся в центре потока, не соприкасаются с эндотелием и потому исключены из процесса обмена веществ. Избыточность капиллярного кровенаполнения и скорости капиллярного кровотока предотвращается констрикцией прекапиллярных сфинктеров, артериол и артерий мышечного типа. При включении в патологический процесс многих органов и тканей вазоспазм артерий МЦР может привести к значительному повышению давления в крупных артериях и перегрузке сердца, что чревато осложнениями и для МЦР, поэтому существует целая система механизмов вазодилатации, которая вместе с вазоконстрикторными реакциями позволяет до определенного предела сохранять баланс «интересов» капилляров, с одной стороны, и более крупных структур сердечно-сосудистой системы – с другой.
Во-вторых, следует иметь в виду, что при любых процессах, происходящих в МКК, в капилляры системы легочной артерии поступает не артериальная, а венозная кровь с очень низким содержанием кислорода. Ввиду этого через аэрогематический барьер, состоящий из слоя капиллярных эндотелиоцитов, двух тончайших базальных мембран и одного слоя альвеолоцитов, диффузия кислорода происходит не из капилляра, как в БКК, а в капилляр из богатого кислородом воздуха, находящегося в просвете альвеол. И именно этот кислород обеспечивает энергетические нужды всех структурных элементов стенок альвеол и альвеолярных ходов, в том числе микрососудов, входящих в их состав. Следовательно, при изучении МКК важно оценивать не только гемодинамические критерии, но и параметры респираторной функции легких. Что касается интерстициальных структур легких и входящих в их состав сосудов, бронхов и нервов, то они обеспечиваются артериальной кровью из системы бронхиальных артерий БКК, а также из венул МКК.
Избыточное количество крови, поступающее в МКК, оказывает повышенное давление на стенки артерий. Усиление трансмурального давления, как уже указывалось, является одним из главных сигналов, включающих синтез эндотелиоцитами резистивных артерий вазоспастических веществ.
Для МКК наиболее существенными из изученных ныне эндогенных вазоконстрикторов следует считать эндотелин и вазопрессин, так как к ангиотензину II, которым легкие обеспечивают весь БКК, на гладкомышечных клетках артерий МКК отсутствуют специфические рецепторы.
Эндотелийзависимые вазопрессоры вызывают спазм артерий мышечного типа с сокращением просвета, что особенно выражено в наиболее мелких сосудах.
Сокращение артериального просвета при неизмененном систолическом кровенаполнении МКК приводит к усилению действия на эндотелий напряжения сдвига крови, что запускает синтез эндо- телиоцитами очень мощного, но короткоживущего вазодилататора NO, который на непродолжительное время снимает спазм артерий. Однако до тех пор, пока не будет устранен ВПС, приведший к перегрузке легочной артерии, спазм артерий МЦР, направленный на поддержание оптимального капиллярного кровотока в стенках альвеол, будет постоянно возобновляться. В медии артерий гиперфункция гладкомышечных клеток приводит к их гипертрофии, в первую очередь это касается мелких артерий. При этом, гладкомышечные волокна распространяются по МЦР в дистальном направлении. Артериолы приобретают строение артерий с несколькими полноценными слоями мышечных волокон в медии. Иммуногистохимическая реакция с моноклональными антителами, специфичными к актину гладкомышечных клеток, позволяют выявить последние даже в межальвеолярных перегородках, где они в норме всегда отсутствуют. Это свидетельствует о значительном увеличении мышечного компонента на уровне прекапиллярных сфинктеров.
В более крупных артериях гипертрофия медии сопровождается гиперэластозом.
Одновременно с этим начинают происходить изменения внутреннего слоя артерий. Хроническое гемодинамическое травмирование интимы приводит к ее концентрическому или подушкообразному утолщению за счет фибромышечной пролиферации.
В наиболее крупных, деформированных внутридолевых артериях фибромышечная пролиферация интимы больше всего выражена в зонах изгибов сосудов, где ламинарный кровоток превращается в турбулентный. Со временем компенсаторная гипертрофия медии сменяется нарастающим фиброзом резистивных сосудов. Из структур, призванных регулировать периферический кровоток, они превращаются в ригидные трубки, слабо отвечающие на изменения центральной гемодинамики. При этом многие мелкие артерии и артериолы частично или полностью облитерируются как за счет концентрической гипертрофии-гиперплазии, так и за счет микротромбов, формирующихся на интиме с поврежденным эндотелием.
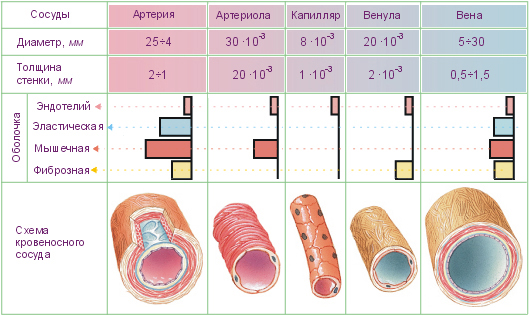
Блок внутридольковых артерий, с одной стороны, выключает из кровотока часть капилляров и приводит к мелкоочаговому фиброзу легочной ткани, с другой – вызывает повышение АД в проксимальных сосудах. Последние отвечают на это деформацией, особенно выраженной в более крупных артериях, окруженных достаточно широкими прослойками рыхлой волокнистой соединительной ткани. Артерии приобретают змеевидную форму. На последовательных серийных гистологических срезах можно наблюдать, что стороны изгибов часто бывают соединены своеобразными «обводными каналами» в виде артерий гораздо меньшего калибра, чем «материнская» артерия и ветви, на которые последняя дихотомически делится в дистальном направлении. Такие изменения крупных артерий увеличивают суммарный объем проксимальной части МКК и частично способствуют снижению сопротивления правому желудочку сердца.
Однако главной мерой, призванной разгрузить правый желудочек, является система артериовенозных анастомозов, бурно развивающаяся при ВПС с прекапиллярной ЛГ. Различают два вида АВА: простые и гломусные. Первые из них обеспечивают постоянное артериовенозное сообщение.
Гломусные АВА снабжены специальными клетками, относящимися к APUD-системе. В одной фазе они синтезируют вазоактивные вещества, при этом сами увеличиваются в объеме и перекрывают многочисленные просветы, выстланные эндотелием. В следующей фазе вазоактивные вещества выходят за пределы гломусных клеток, сокращая их объем и открывая артерио-венозное соустье, вызывая вместе с тем спазм артериального компонента АВА. Кровь поступает в вену, разгружая артерии и правый желудочек, но при этом обкрадывая капиллярный кровоток. Это вместе с редукцией части капилляров в результате описанных выше изменений мелких артерий вызывает или усугубляет общую гипоксию организма, связанную непосредственно с ВПС. Любая гипоксия компенсируется форсированным дыханием, усиливающимся при минимальной физической нагрузке и других неблагоприятных обстоятельствах. При форсированном дыхании повышается внутриальвеолярное давление, которое может превышать давление в капиллярах со сниженным кровотоком, полностью или частично блокируя последний, что, в свою очередь, усугубляет общую гипоксию и усиливает респираторный компонент патогенеза ЛГ. Хроническое повышение внутриальвеолярного давления приводит к расширению просветов альвеол и альвеолярных ходов. В результате этого, как при эмфиземе любого генеза, по периферии альвеол, у их стенок формируется так называемая мертвая зона, где воздух не меняется в процессе дыхания и, таким образом, прекращается диффузия кислорода из воздуха в кровь капилляров МКК. Это усугубляет общую гипоксию организма и приводит к гипоксическому поражению структур альвеолярной ткани легких, включая капилляры и артериолы. Многие межальвеолярные перегородки полностью атрофируются, другие оказываются лишенными капилляров и представлены в основном коллагеновыми волокнами.
Свою лепту в патогенез ЛГ вносят воспалительные и аллергические заболевания легких, которыми часто страдают больные с ВПС. Рецидивирующие бронхиты и пневмонии разрешаются фиброзом, замещающим определенные участки легочной ткани, с редукцией компонентов аэрогематического барьера. Обструктивные бронхиты и бронхиальная астма сопряжены с гиповентиляцией альвеол, которая, как уже говорилось, вызывает спазм артерий гиповентилируемых зон, что усугубляет гемодинамический компонент патогенеза ЛГ, завершающегося редукцией капиллярной сети альвеол. Очевидно, что степень суммарной редукции капиллярного русла обоих легких является тем интегративным показателем, который может дать представление о глубине трансформации МКК при перегрузке его объемом, а затем и давлением.
Пороки сердца, сопряженные с затруднением оттока крови из МКК по легочным венам способствуют развитию посткапиллярной ЛГ. Последняя возникает из-за перегрузки легочных вен в результате их неполного освобождения при систоле предсердий. Мелкие вены и венулы, физиологически играющие роль резервных емкостей для кровеносной системы, значительно расширяются, увеличивая суммарный объем венозной сети и таким образом до определенной степени компенсируя повышение венозного давления в МКК. Однако увеличение кровенаполнения венул закономерно передается на капилляры и дистальные элементы артериального русла легких. В ответ на это спазмируются прекапиллярные сфинктеры, артериолы и мелкие артерии. Поступление крови в капилляры снижается, но из-за низкого градиента давлений между артериальным и венозным концами капилляров скорость кровотока в них замедляется, что значительно снижает качество аэрогематического газообмена и способствует развитию гипоксии. Кроме того, постоянное спазмирование мелких артерий, прерываемое лишь действием NO, вызывает их ремоделирование, так же как при прекапиллярной форме ЛГ. Это ведет к деформации более крупных артерий с образованием описанных выше артерио-артериальных шунтов. Однако ЛВД в легких больных с посткапиллярной гипертензией встречаются очень редко и только на поздних стадиях развития болезни. По-видимому, их образованию препятствует высокое давление в легочных венах.
В дальнейшем после редукции просветов многих мелких артерий у больных данной категории изменения структур легких происходят такие же, как и при ВПС с прекапиллярной ЛГ. При этом гипоксическое повреждение стенок сосудов МЦР в сочетании с избыточным трансмуральным давлением настолько повышает проницаемость аэрогематического барьера, что он в критические моменты начинает пропускать жидкую часть крови и даже эритроциты. Последние лизируются, гемоглобин поглощается альвеолярными макрофагами, которые в таком состоянии называются гемосидерофагами, или клетками сердечных пороков. Именно они окрашивают мокроту больных в бурый цвет. Гемосидерофаги иногда заполняют значительную часть просветов альвеол и поэтому способствуют развитию гипоксии, которая так же, как и при прекапиллярной гипертензии, включает респираторный компонент патогенеза ЛГ.
Таким образом, в патогенезе пре- и посткапиллярной ЛГ, имеются как общие, так и отличительные черты. Общие – ремоделирование артерий, редукция капиллярного русла и участие респираторного механизма в формировании изменений, первично индуцированных нарушениями центральной гемодинамики. Различия заключаются в том, что при исходном высоком давлении в легочных венах на разных этапах патогенеза все сохраняющиеся капилляры МКК остаются доступными для перфузии, тогда как АВА, формирующиеся при ВПС с перегрузкой легочной артерии, «обкрадывают» и без того редуцирующееся капиллярное русло, способствуя нарастанию этой редукции. В связи с этим критические состояния больного, обусловленные эпизодами сердечной слабости, повышением физической нагрузки и другими факторами, при прекапиллярной ЛГ сопровождаются одышечно-цианотическими приступами, тогда как для посткапиллярной ЛГ более характерно развитие отека легких.
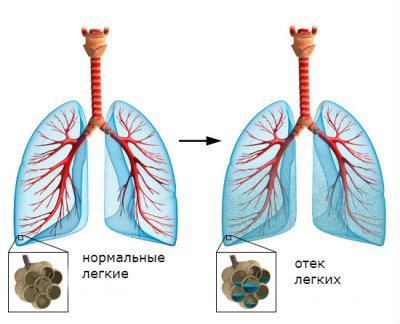
Правильное понимание механизмов развития ЛГ может быть полезным, как для ведения каждого конкретного больного, так и для усовершенствования схем диагностики и лечения этого серьезного осложнения многих ВПС.
Субклеточные механизмы формирования структурных изменений при ЛГ
Процессы, сопровождающие нормальное развитие структуры легочных сосудов, вовлечены также в развитие ЛГ:
-
дистальное распространение гладкой мускулатуры;
-
гипертрофия медии и адвентиции;
-
увеличение продукции белков матрикса в стенках артерий.
Прогрессирование этих изменений коррелирует с постепенной потерей способности к вазодилатации.
Соединительнотканные элементы сосудистой стенки представлены эластином, коллагенами, протеогликанами и гликопротеинами. Легочные сосуды новорожденного активнее отвечают на гипоксию и гемодинамические стрессы клеточной пролиферацией, синтезом протеинов матрикса, накоплением эластина и коллагена, чем сосуды взрослого. Таким образом, структурные изменения легочных сосудов у детей младенческого возраста развиваются очень быстро.
Легочная гипертензия сопровождается изменениями фенотипа гладкомышечных клеток и появлением субпопуляций клеток, имеющих различную способность к пролиферации и продукции белков матрикса.
-
Эндотелиальная дисфункция.
Эндотелий легочных сосудов играет решающую роль не только в регуляции сосудистого тонуса в норме, но и в патогенезе ЛГ. Повреждение эндотелия, происходящее вследствие гидродинамических нагрузок, вызывает нарушения нормальной эндокринной функции эндотелиальных клеток.
В последние годы стало очевидным, что повреждение эндотелия приводит к нарушению реактивности легочных сосудов и пролиферации гладкомышечных клеток легочных сосудов и фибробластов. Кроме этого, эндотелий принимает участие в свертывании крови, продукции цитокинов и факторов роста.
Нарушение механизма образования NО является одним из важных элементов патогенеза ЛГ. Поскольку освобождение NО стимулируется факторами эндотелиального происхождения, такими, как ацетилхолин, последний широко применяется в исследованиях степени повреждения эндотелия. Эндотелиальная дисфункция и снижение продукции NО, которые оцениваются по степени вазодилатации в ответ на ацетилхолин, обнаружены после искусственного кровообращения и у больных с ВПС, осложненными ЛГ. В частности, обнаружен его недостаток в гипертензивных легочных артериях при синдроме Эйзенменгера.
Современными исследованиями установлено, что эритроциты играют важную роль в поддержании нормальной микроциркуляции в легких: продвигаясь по капиллярам, они деформируются, этот процесс сопряжен с генерацией эритроцитами вазоактивных субстанций, которые, взаимодействуя с рецепторами клеток эндотелия сосудов, вызывают расслабление последних. Показано, что эритроциты больных с ВПС, осложненными ЛГ, теряют способность к деформации. Известно, что NО является одним из регуляторов деформируемости эритроцитов. Существование в эритроцитах собственной NО-синтазной системы генерации NО при наличии гемоглобина, способного восстанавливать NО из нитритов и нитратов, дает возможность предположить, что NО является одним из важных регуляторов функционального состояния эритроцитов. Кроме того, исследования, проведенные на линии спонтанно гипертензивных крыс, показали, что вызванные гипертензией изменения в системе NО, происходящие в эритроцитах, аналогичны тем, которые происходят в эндотелии сосудов.
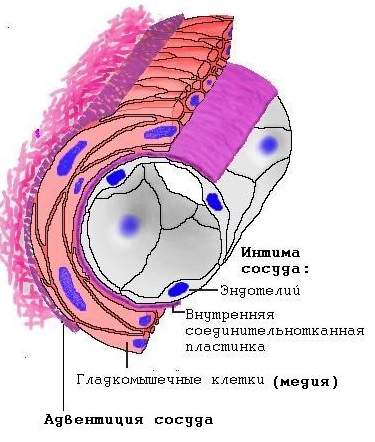
Отечественные исследования проблемы
Принимая во внимание вышеизложенные данные литературы относительно эритроцитов, а также учитывая большую доступность этих клеток крови для проведения исследований по сравнению с клетками эндотелия сосудов, отечественные специалисты совместно с Гулой и Косяковой изучали активность системы генерации NO и ее вклад в поддержание сосудистого тонуса у больных с ДМЖП и АВСД с различными стадиями ОБЛС и давленим в легочной артерии, приближающимся к системному или равным ему.
Возраст пациентов колебался от 3 мес до 48 мес. Были выделены 3 клинико-гемодинамические группы. Первую группу составили пациенты с застойной сердечной недостаточностью и индексом Wood, не превышающим 5 ед./м2. После коррекции порока давление в легочной артерии нормализовалось. Во вторую группу вошли пациенты с признаками повышенного ЛСС и KW 6-8 ед./м2. После операции давление в легочной артерии без применения вазодилататоров снизилось и составило до 50-60% системного. В третью группу включены больные с выраженной ОБЛС и компромиссными показаниями к операции: KW 9,8-12,3 ед./м2. Давление в легочной артерии не снизилось или снизилось незначительно. В контрольной группе были пациенты с теми же диагнозами, но с нормальным давлением в легочной артерии.
Изучали активность сNOS и iNOS в эритроцитах артериальной и венозной крови, а также содержание стабильных метаболитов NO в эритроцитах и плазме крови. Результаты исследований активности сNOS в эритроцитах артериальной и венозной крови контрольной группы пациентов показали, что в эритроцитах артериальной крови активность фермента выше по сравнению с таковой в эритроцитах венозной крови, что, скорее всего, вызвано разницей в парциальном давлении кислорода, а кислород является одним из субстратов для NO-синтазной реакции. Обнаружено, что у больных с ЛГ по мере прогрессирования ОБЛС происходит постепенное снижение активности фермента по сравнению с пациентами контрольной группы в эритроцитах как артериальной, так и венозной крови.
Как видно из графика, в эритроцитах артериальной крови пациентов контрольной группы активность сNOS до операции в 2-2,5 раза выше активности индуцибельной изоформы, что указывает на то, что поддержание нормального тонуса сосудов обеспечивается главным образом за счет сNOS. У пациентов с ЛГ происходит изменение в соотношении изоформ NOS: активация индуцибельной изоформы, которая возникает вследствие адаптации к увеличенному легочному кровотоку и способствует образованию большего количества NO в сравнении с той, которая генерируется в результате работы сNOS. По мере прогрессирования болезни iNOS вызывает ингибирование конститутивной изоформы вследствие конкуренции за субстрат L-аргинин.
На начальной стадии ЛГ активность iNOS возрастает, что является, адаптивной реакцией на увеличенный кровоток, поскольку благодаря iNOS за короткий период времени генерируется значительно большее количество NO в сравнении с тем, которое образуется конститутивной изоформой фермента. Возрастание активности iNOS сопровождается соответствующим снижением активности cNOS вследствие конкуренции за субстрат L-аргинин.
Обращает на себя внимание снижение активности обеих изоформ NOS у пациентов II группы в сравнении с больными I группы. Такое снижение может быть вызвано исчерпанием пула L-аргинина, что является характерным для развития гипертензии. Вероятно, состояние гемодинамики на этом этапе является переломным в развитии патологического процесса и свидетельствует об истощении адаптивных возможностей организма и углублении патологических нарушений.
У пациентов III группы отмечается дальнейшее снижение активности cNOS на фоне гиперактивации iNOS. Источником субстрата для iNOS в этом случае может выступать L-аргинин, который выходит из клеток эндотелия в результате деструктивных изменений, характерных для этой стадии заболевания. Известно, что развитие ЛГ связано с процессами деэндотелизации сосудов. Значительное повышение активности iNOS в эритроцитах пациентов III группы указывает на развитие воспалительных процессов и оксидативного стресса. Активация iNOS приводит к генерации избыточных количеств NO, которые принимают участие в свободнорадикальных процессах повреждения сосудов. Поэтому вызванная адаптивными реакциями на гиперволемию активация iNOS в начале заболевания превращается в мощный повреждающий фактор на поздних его стадиях. Предыдущие исследования экспрессии сNOS и iNOS в легочной ткани больных со вторичной ЛГ показали возрастание экспрессии именно iNOS. Таким образом, исходя из результатов наших исследований, можно сказать, что эритроциты отражают те изменения, которые происходят непосредственно в легочной ткани.
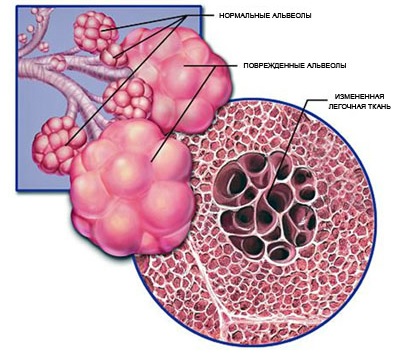
Вследствие активации iNOS в эритроцитах артериальной и венозной крови больных с ЛГ возрастает содержание стабильных метаболитов NO. Получен данные об увеличении содержания стабильных метаболитов NO по мере прогрессирования ОБЛС коррелируют с другими данными для плазмы крови, а также данными литературы.
В последнее время сложилось представление о двоякой роли NO в патогенезе хронической ЛГ. Komai и соавторы изучали экспрессию сNOS в биоптатах легочной ткани у детей с ВПС и ЛГ с помощью моноклональных антител против сNOS. Они обнаружили положительную связь между уровнем экспрессии сNOS и стадией ОБЛС. Takay и соавторы продемонстрировали наличие связи между концентрацией стабильных метаболитов NO в плазме и средним давлением в легочной артерии у детей с ДМЖП.
Усиленный синтез эндогенного вазодилататора NO является адаптивной реакцией, противодействующей чрезмерному повышению давления в легочной артерии в момент острой вазоконстрикции и хронической ЛГ, хотя было также описано снижение активности NOS при ЛГ. Как показано выше, специалисты обнаружили активацию окислительного метаболизма L-аргинина, зависимость ее от стадии ОБЛС и изменение соотношения активности изоформ NOS, что позволяет объяснить противоречивость данных литературы. Оксид азота может способствовать повреждению сосудов с последующим фиброзом. С одной стороны, NO предупреждает повреждение сосудов кислородными радикалами, которые при ЛГ продуцируются в повышенных количествах, и тем самым препятствует пролиферации гладкомышечных клеток и фибробластов. С другой стороны, NO, вступая в реакцию с кислородными радикалами, образует пероксинитрит и другие высокоагрессивные соединения. Эти оксиданты оказывают цитотоксическое и коллагенолитическое действие, способствуя процессу репаративного ремоделирования сосудов. Баланс между защитным и повреждающим эффектами NO определяется соотношением количества NO и активных кислородных радикалов. Он может сместиться в сторону более тяжелого повреждения, особенно при хронических заболеваниях, сопровождающихся ЛГ. Кроме того, показано, что утрата эритроцитами способности к деформации может быть вызвана как недостатком, так и избытком NO. Таким образом, изменение функционального состояния эритроцитов под влиянием высоких концентраций NO может значительно сказываться на состоянии легочной микроциркуляции.
Аминокислота L-аргинин подвергается также неокислительному метаболизму, являясь субстратом и для другого фермента – аргиназы. Последняя осуществляет гидролиз L-аргинина с образованием L-орнитина и мочевины. L-орнитин, в свою очередь, может подвергаться декарбоксилированию с помощью фермента орнитиндекарбоксилазы с образованием путресцина – предшественника синтеза полиаминов. Таким образом, аминокислота L-аргинин является субстратом для двух ферментативных реакций – NO-синтазной и аргиназной, которые, соответственно, конкурируют за субстрат.
Отечественные исследования уровня полиаминов у пациентов с ЛГ до и через 12 ч после операции с искусственным кровообращением показали, что для контрольной группы характерны низкие уровни суммарных полиаминов, тогда как во всех группах больных с ЛГ происходит постепенное возрастание их содержания в эритроцитах в зависимости от стадии ОБЛС. Наиболее высокие уровни полиаминов выявлены в эритроцитах пациентов III группы, т.е. при выраженных структурных изменениях легочных сосудов.
Полиамины являются продуктами анаболического звена метаболизма и синтезируется в результате декарбоксилирования аминокислот. Представители этой группы соединений служат мощными факторами роста и пролиферации. Исследованиями последних лет установлено, что полиамины принимают участие в регуляции тонуса сосудов, вызывая, в зависимости от их концентрации, констрикторные или дилататорные реакции гладкой мускулатуры сосудов. Кроме того, показана способность всех представителей полиаминов к ингибированию NО-синтаз.
Данные литературы свидетельствуют о накоплении полиаминов в легких и в эндотелии легочных сосудов под влиянием гипоксии, что, по мнению исследователей, связано с активацией в этих условиях процессов транспорта полиаминов, а не с увеличеним их синтеза.
Для эритроцитов не установлена способность к синтезу полиаминов de novo, однако доказано их участие в транспортировке последних. Следовательно, значительное увеличение содержания полиаминов в эритроцитах больных свидетельствуют об интенсивности пролиферативных процессов в тканях легких и сосудах, которые обусловливают утолщение интимы сосудов, вследствие чего уменьшается их просвет. Таким образом, увеличение содержания полиаминов способствует развитию гипертензивных осложнений у пациентов с ВПС.
Обнаруженная отечественными кардиологами тесная связь содержания суммарных полиаминов в эритроцитах со стадией ОБЛС позволяет использовать этот факт в качестве дополнительного диагностического метода для оценки операбельности больных в пограничных случаях, когда клинические и гемодинамические показатели неоднозначны.
Эндотелин-1 играет важную роль в механизме повышения ЛСС при различных состояниях, связанных с ЛГ, например:
-
при хронической гипоксии;
-
синдроме Эйзенменгера;
-
первичной ЛГ;
-
ВПС, сопровождающихся ЛГ;
-
персистирующей ЛГ новорожденных;
-
на высокогорье.
При этом вазоконстрикторные эффекты ЭТ-1 преобладают над сосудорасширяющими. В патогенезе легочного гипертензивного статуса определяющим является дисбаланс между системами NO-цГМФ и ЭТ. Снижение продукции NO может привести к увеличению выработки ЭТ-1, который, являясь мощным митогеном, стимулирует пролиферацию гладкой мускулатуры и фибробластов.
-
Эйкозаноиды.
У детей с ЛГ обнаружен дисбаланс в биосинтезе эйкозаноидов в пользу тромбоксана, и, следовательно, в пользу вазоконстрикции и проагрегации. Так, при ДМЖП с ЛГ активируются тромбоциты в легочных капиллярах. Это проявляется повышением уровня стабильного метаболита томбоксана А2 – тромбоксана В2. Связь концентрации тромбоксана В2 с давлением в легочной артерии выражается логарифмической зависимостью. Дисбаланс в биосинтезе эйкозаноидов отмечается на ранней стадии развития болезни легочных сосудов у младенцев с потенциально обратимыми изменениями сосудов, а также у больных в юношеском возрасте с выраженными необратимыми изменениями легочных сосудов. Это происходит в основном за счет снижения способности поврежденного эндотелия продуцировать простациклин. Таким образом, эйкозаноиды играют важную роль в развитии ЛГ.
Повреждение эндотелиальных клеток является причиной увеличения продукции протромботических эндотелиальных факторов. Тромбоз сосудов – одна из причин развития первичной ЛГ, а антикоагуляция увеличивает выживаемость больных и способствует улучшению легочной гемодинамики у взрослых больных с ЛГ. Баланс между фибринолитической активностью активатора тканевого плазминогена и антифибринолитической активностью ингибитора активатора плазминогена I типа при ЛГ нарушен, что указывает на роль эндотелиальной дисфункции в патогенезе ЛГ. Уровень ИАП-1 в плазме при ЛГ повышен. При ВПС, а также при первичной ЛГ увеличена эндотелиальная продукция фактора von Willebrand, что также вносит вклад в предрасположенность к тромбообразованию при ЛГ.
Среди причин структурных изменений легочных сосудов при гипертензии определенная роль принадлежит факторам роста – гладкомышечным митогенам, влияющим на подлежащие гладкомышечные клетки, и их способности к дистальной миграции и пролиферации. Очевидную роль в развитии ЛГ играет увеличение активности протеолитических ферментов, особенно эндогенной сосудистой эластазы. ЭСЭ была идентифицирована как сывороточная протеаза адипсин. Первичные гемодинамические изменения стимулируют продукцию ЭСЭ, которая способствует миграции гладкой мускулатуры и пролиферации. Важную роль ЭСЭ демонстрирует смягчение синдрома ЛГ в ответ на блокаду сывороточных эластаз.
При ЛГ увеличена активность фибробластов. Пролиферация фибробластов предшествует пролиферации гладкомышечных клеток, что указывает на их роль в изменениях легочных сосудов при гипертензии. Кроме того, она может препятствовать нормальной фенотипической дифференциации протеинов матрикса.
В некоторых экспериментальных моделях ЛГ обнаружен феномен удлинения времени релаксации гладкомышечных клеток, что вносит дополнительный элемент в механизмы ее развития. В других исследованиях установлено снижение уровня фосфатазы – фермента, ответственного за релаксацию гладкой мускулатуры.
Патофизиология ЛГ
Легочная гипертензия, вызванная у младенцев внезапной обструкцией верхних дыхательных путей или массивной тромбоэмболией легочных артерий, развивается быстро и в условиях не гипертрофированного правого желудочка приводит к острой правожелудочковой недостаточности. При хронической ЛГ постепенно развиваются гипертрофия и дилатация правого желудочка. Давление в нем может превышать системное, если отсутствует сообщение на уровне межжелудочковой перегородки. Сердечный выброс снижается. В основе этого лежат, по крайней мере, два механизма. Прежде всего, перегрузка правого желудочка давлением и объемом ухудшает функцию сердца за счет снижения коронарной перфузии гипертрофированного правого желудочка и ухудшения функции левого желудочка из-за выбухания межжелудочковой перегородки влево. Смещение перегородки нарушает структуры левого желудочка и снижает его комплайнс, за чем следует повышение конечно-диастолического давления в левом желудочке и увеличение давления в левом предсердии. Второй механизм состоит в снижении притока легочной венозной крови в левое предсердие, что приводит к гипотензии и циркуляторному шоку при отсутствии право-левого внутрисердечного шунта.
У детей может развиться отек легких даже без повышения давления в левом предсердии вследствие разрыва стенок артериол, расположенных проксимальнее артериол, сузившихся в результате гипоксической вазоконстрикции. Этот механизм аналогичен таковому при отеке легких на высокогорье. Надрывы происходят в артериолах, в которых отсутствует гипертрофия гладкой мускулатуры медии. Насыщение артериальной крови кислородом снижается. В результате легочного венозного застоя или отека, сдавления дыхательных путей или внутрисердечных шунтов развиваются гипоксемия, ацидоз и даже гиперкапния.








- Аллергия
- Ангиология
- Болезни глаз
- Венерология
- Гастроэнтерология
- Гинекология
- Дерматология
- Здоровое питание
- Инфекционные болезни
- Кардиология
- Косметология
- Лекарства
- Лекарственные растения
- ЛОР-заболевания
- Мужское здоровье
- Неврология
- Неотложная помощь
- Новости
- Онкология
- Ортопедия
- Паразитология
- Педиатрия
- Пульмонология
- Расшифровка анализов
- Симптомы
- Системные заболевания
- Стоматология
- Травматология
- Урология
- Хирургия
- Эндокринология
- Нужно знать
- Еда
- Профессиональные заболевания
Комментарии