
Порок клапана легочной артерии. Врожденный порок КЛА
Врожденное отсутствие легочного клапана является относительно редкой патологией. Синдром тетрады Фалло с отсутствующим клапаном легочной артерии встречается у 2-6% пациентов с тетрадой Фалло. В большинстве случаев эта аномалия сопровождается ДМЖП, сужением кольца легочного клапана и резкой дилатацией легочных артерий. Эту комбинацию аномалий часто называют «тетрадой Фалло с отсутствующим легочным клапаном». Существует также изолированное отсутствие легочного клапана с интактной межжелудочковой перегородкой, которое встречается еще реже. После первого описания этого порока в 1847 г. Chevers опубликовано более 300 подобных наблюдений, в том числе 15 — с интактной межжелудочковой перегородкой. Отличительными особенностями этого синдрома являются:
-
отсутствующие или рудиментарные створки клапана;
-
аневризматическая дилатация одной или обеих легочных артерий, сдавливающих бронхи;
-
врожденная агенезия артериального протока, при сливающихся легочных артериях.
Эмбриология и генетика
При этом синдроме часто обнаруживали делецию хромосомы 22. Таким образом, существует специфический эмбриональный механизм, включающий взаимодействие неврального гребешка и примитивных аортальных дуг.
Существует предположение, что резкая дилатация легочных артерий у плода патогенетически связана с агенезией артериального протока. Это сочетание было воспроизведено в эксперименте у плодов крыс путем введения тератогена бис-диамина. Отсутствие легочного клапана было описано у больного с интерстициальной делецией длинной ветви хромосомы 6. Отсутствие легочного клапана наблюдается при синдроме Di George.
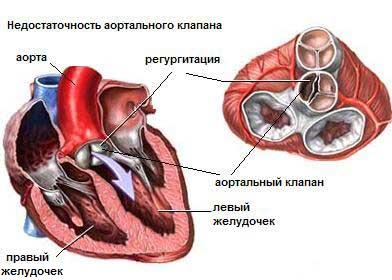
Анатомия
В большинстве случаев отсутствует зрелый легочный клапан. Рудиментарный легочный клапан состоит из бессосудистых масс, выдающихся в просвет легочной артерии. На месте предполагаемого клапана обнаруживаются участки тонкой невыраженной примитивной узловатой желатинообразной соединительной ткани. У некоторых больных имеется неполная и несовершенная, но зрелая ткань клапана. Прогноз в этих случаях относительно хороший. Пациенты часто переживают период новорожденности, однако многие младенцы с тяжелой формой этого синдрома умирают рано от дыхательной и толерантной к лечению сердечной недостаточности.
Сердце обычно сильно увеличено за счет правого желудочка и его инфундибулярного отдела. Легочная артерия аневризматически дилатирована. Кольцо клапана гипоплазировано, инфундибулум узкий, длинный и извитой.
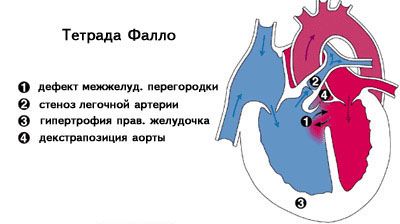
При наличии ДМЖП и инфундибулярного стеноза внутренняя анатомия сердца соответствует таковой при тетраде Фалло. У этих пациентов имеются все анатомические признаки тетрады Фалло, в том числе у 25% — правосторонняя дуга аорты. В данном варианте тетрады Фалло встречаются типичные аномалии коронарных артерий. Легочные артерии могут быть не связанными между собой и кровоснабжаться через боталлов проток или отходить непосредственно от восходящей аорты. Дефект межжелудочковой перегородки большой, расположен в области мембранозной перегородки ниже аортального клапана. Восходящая аорта расширена.
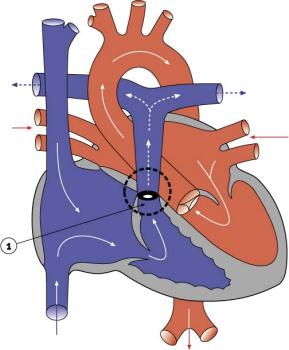
Стенка дилатированной легочной артерии патологически изменена и в отдельных случаях гистологически напоминает таковую при синдроме Марфана, однако у большинства пациентов не обнаружены аномалии ее архитектуры.
Существует взаимосвязь между ориентацией инфундибулума и степенью дилатации легочной артерии. Если конус правого желудочка ориентирован вправо, правая легочная артерия аневризматически расширена, в то время как вертикальная и левосторонняя ориентация инфундибулума сопровождается двусторонней дилатацией или расширением левой легочной артерии. В патогенезе дилатации легочной артерии определенную роль играет гипоплазия легочного кольца, приводящая к постстенотическому расширению.
Легочные проблемы обусловлены сдавлением трахеобронхиального дерева дилатированными легочными артериями и аномально отходящими мелкими легочными артериальными сосудами, которые образуют сосудистые пучки, обвивающие и сдавливающие мелкие внутрилегочные бронхи. В некоторых случаях количество альвеолярных разветвлений бронхов значительно уменьшено, в связи с чем, у пациентов с тяжелой формой синдрома хирургическое устранение компрессии главных бронхов не всегда облегчает состояние и не избавляет от вероятности рецидива обструкции дыхательных путей.
Могут иметь место аортолегочные коллатерали различных размеров. Чаще они малые и непрямые, реже бывают большими и прямыми. Коллатерали отходят от нисходящей грудной аорты и входят в корень легкого. Они осложняют хирургическое вмешательство, если не обработаны перед коррекцией.
Порок сочетается с ОАП, ДМПП, отхождением магистральных артерий от правого желудочка, синдромом Марфана, ДМЖП без стеноза клапанного кольца, АТК, ТМА, аномалией Uhl, отхождением правой или левой легочной артерии от восходящей аорты, коарктацией аорты. Данный синдром могут сопровождать также аортальный стеноз, полный или частичный АВСД, ТАДЛВ, azygos продолжение нижней полой вены и др..
Клиника
Отсутствие клапана легочной артерии можно заподозрить по клиническим признакам. Грубый систолодиастолический шум у цианотичного новорожденного с сопутствующей дыхательной недостаточностью обычно указывает на наличие тетрады Фалло с отсутствующим легочным клапаном.
У большинства пациентов с тяжелой формой порока сразу после рождения развивается респираторный дистресс-синдром. Цианоз отсутствует, за исключением раннего неонатального периода. Дыхательная недостаточность обусловлена частичным сдавлением бронхов резко расширенными легочными артериями и компрессией внутрилегочных бронхов аномально ветвящимися дистальными ветвями легочных артерий. Дыхание улучшается, когда ребенка кладут навзничь. Тяжелая сердечная недостаточность скоро приводит к смерти. При аускультации обнаруживается характерный систолодиастолический шум «шум хрустящего под ногами снега». При наличии ДМЖП наблюдается умеренный цианоз, частое абдоминальное и глубокое грудное дыхание с втяжением межреберных промежутков. Часто отмечается стридор на вдохе и выдохе. По левому краю грудины определяется систолическое и иногда диастолическое дрожание. Систолодиастолический шум лучше слышен сверху по левому краю грудины и широко распространяется над всей предсердечной областью. Шум состоит из грубого систолического компонента, обусловленного сужением кольца легочного клапана, и грубого убывающего диастолического компонента. Шум прерывается нерасщепленным II тоном, который лучше слышен внизу по нижнему краю грудины и на верхушке. Он генерируется закрывающимся аортальным клапаном. Прерывистость систолодиастолического шума отличает его от типичного непрерывного шума ОАП.
Вследствие обструктивной природы дыхательной недостаточности увеличение печени наблюдается редко. Однако у некоторых пациентов с выраженной сердечной недостаточностью печень может быть значительно увеличенной. У больных без явной сиптоматики II сердечный тон может быть расщеплен, что указывает на наличие функционирующего клапана. В этих случаях на вскрытии обнаруживают двустворчатый легочный клапан. У пациентов, переживших первый год жизни, симптомы дыхательной недостаточности спонтанно уменьшаются, исчезает цианоз. Синюшность исчезает при реверсии шунта вследствие снижения ЛСС, что обычно происходит в неонатальном периоде. При снижении сосудистого сопротивления уменьшаются недостаточность клапана легочной артерии, диастолическая перегрузка правого желудочка и право-левый сброс на желудочковом уровне во время диастолы. Небольшой шунт крови справа налево во время диастолы может сохраняться даже при умеренном систолическом сбросе слева направо.
Электрокардиография
Для тетрады Фалло и отсутствия клапана легочной артерии характерна гипертрофия правого желудочка. При наличии ДМЖП, перекрестного или лево-правого шунта может регистрироваться гипертрофия обоих желудочков. Изолированная недостаточность клапана на ЭКГ проявляется невыраженной гипертрофией правого желудочка, которую можно заподозрить по rsR' в правых отведениях.
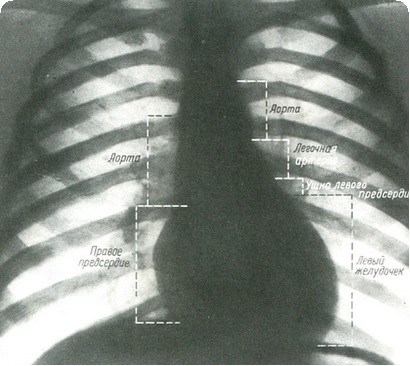
Рентгенография
На обзорной рентгенограмме умеренно увеличенное сердце с резко расширенными стволом и ветвями легочной артерии. Однако при правосторонней дуге, сильно раздутом легком со смещением сердца влево аневризматическая дилатация легочных артерий может не быть очевидной. Легочные артерии могут быть расположены медиально и скрыты в тени средостения. Плотные корни напоминают опухоль, но легко идентифицируются на флюороскопии по пульсации, синхронной с сердечными сокращениями. Выбухающая тень по левому верхнему краю сердца образована дилатированным выводным трактом правого желудочка. Это объясняется ротацией сердца влево. Периферический легочный рисунок обычно нормальный и может быть усиленным у старших младенцев и детей с преимущественным лево-правым сбросом. Нередко отмечаются односторонняя обструктивная эмфизема и ателектаз отдельных сегментов со смещением сердца в противоположную сторону.
При изолированной врожденной недостаточности клапана легочной артерии рентгенологическая картина может быть почти нормальной, тем не менее, можно обнаружить усиленную пульсацию артерий и «пляску» корней. При этом дилатация легочных артерий не достигает аневризматических пропорций, типичных для тетрады Фалло с отсутствующим легочным клапаном.
Эхокардиография
Так же, как и при классической форме тетрады Фалло, парастернальный доступ по длинной оси, а также четырехкамерный доступ позволяют увидеть большой ДМЖП и праводеленность аорты. Правый желудочек значительно дилатирован, и часто парадоксальное движение межжелудочковой перегородки является признаком недостаточности легочного клапана. В большинстве случаев можно определить малоподвижный рудиментарный легочный клапан в стенозированном кольце. При парастернальном доступе по короткой оси можно увидеть дилатацию легочного ствола и обеих легочных артерий.
Допплер ЭхоКГ выявляет скоростной антеградный систолический ток через суженное легочное кольцо и ретроградный диастолический ток недостаточности клапана в легочной артерии и выводном тракте правого желудочка. Характер тока крови в легочной артерии может имитировать ОАП, однако ретроградный диастолический кровоток в выводном тракте свидетельствует в пользу недостаточности клапана.

Точному диагнозу мешают смещение средостения из-за перерастяжения легких и искажение анатомии сердца при резкой дилатации легочных артерий, поэтому необходимо найти атипичное положение датчика, чтобы получить надлежащее изображение. Эхокардиографический диагноз этого порока можно поставить до родов. Имеются сообщения, что аномалия может быть причиной водянки плода.
Катетеризация сердца
При наличии дефекта межжелудочковой перегородки и стеноза легочной артерии давление в правом желудочке равно системному с преимущественным право-левым сбросом на желудочковом уровне. При отсутствии значительного сужения в неонатальном периоде ЛСС снижается, падает давление в правом желудочке и шунт приобретает лево-правое направление, хотя систолический градиент давления между правым желудочком и легочной артерией всегда имеется. Диастолическое давление в легочной артерии обычно равно таковому в правом желудочке. Только после многочисленных попыток удается получить точное давление в легочной артерии из-за артефактов, вызванных турбулентностью и тяжелым затруднением дыхания у младенцев. Синюшным детям с дыхательной недостаточностью во время катетеризации введение седативных средств противопоказано из-за опасности остановки дыхания. В этих случаях рекомендуется механическая вентиляция легких.
При изолированной недостаточности легочного клапана давление в правом желудочке и легочной артерии нормальное. Отмечаются небольшой систолический градиент на клапане вследствие высокой скорости кровотока через нормальное легочное кольцо и увеличенный ударный объём правого желудочка. В редких случаях встречается истинный стеноз клапана разной степени выраженности.
Ангиокардиография демонстрирует локализацию и выраженность сужения легочного кольца, ориентацию инфундибулума и дилатацию легочных артерий. Створки клапана не идентифицируются, но на уровне стеноза клапанного кольца можно увидеть дискретный гребень рудиментарного клапана. Инфундибулум и правый желудочек обычно дилатированы. При выраженном стенозе виден сброс контрастного вещества справа налево. Контраст дольше задерживается в правом желудочке из-за недостаточности клапана. Контрастирование легочной артерии позволяет увидеть наличие недостаточности клапана и выраженную дилатацию легочных артерий. В редких случаях одна из ветвей отходит от восходящей аорты.
Конечно-диастолический объём правого желудочка у тяжелых детей с плохим прогнозом существенно больше, чем у детей, переживших неонатальный период. Относительные размеры дилатированной правой легочной артерии коррелируют с частотой осложнений и летальности: чем шире правая легочная артерия, тем хуже исходы естественного течения.
Киноангиография позволяет оценить выраженность аномалии ветвления легочных артерий в корнях легких.
Естественное течение
В мире накоплен большой опыт пренатальной диагностики аномалий конотрункуса, в том числе тетрады Фалло. Представляют интерес исходы фетальных случаев тетрады Фалло с отсутствующим клапаном легочной артерии. Так, из 18 случаев, диагностированных в пренатальном периоде, в 7 случаях семьи приняли решение о прерывании беременности, у 4 плодов наступила спонтанная внутриутробная смерть, 3 детей умерли в неонатальном периоде и 2 — умерли в младенческом возрасте. Таким образом, только 2 детей выжили при сохраненной беременности. В некоторых из этих случаев была диагностирована водянка плода и полноводие. В другой публикации диагноз синдрома был поставлен в 20 случаях, в 6 из них беременность завершена в срок, в 3 случаях наступила внутриутробная смерть, 5 новорожденных умерли, 3 ребенка умерли в младенческом возрасте и 3 пациента остались живыми. Десять из 11, родившихся живыми младенцев нуждались в ранней вентиляции.
Прогноз при этом синдроме зависит от тяжести обструкции трахеобронхиального дерева. Часто причиной смерти являются банальная респираторная инфекция на фоне обструктивной эмфиземы и ателектазы. В легких случаях состояние спонтанно улучшается к 6-9 мес первого года жизни. Это происходит благодаря созреванию структуры бронхов и снижению давления в легочных артериях. У новорожденных нормальной особенностью трахеобронхиального дерева является слабость хрящевой, мышечной и эластической ткани трахеи и бронхов. В процессе созревания бронхи становятся более жесткими и устойчивыми к внешнему давлению. Зрелые бронхи становятся более широкими, поэтому внешнее давление создаёт меньшую обструкцию. Снижение ЛСС и прогрессирование сужения клапана приводят к уменьшению давления в легочной артерии и к меньшему растяжению дилатированых легочных артерий.

Только изредка пациенты с этим синдромом обнаруживаются среди взрослых людей. У некоторых больных клиническая картина его такая же, как у пациентов с обычной тетрадой Фалло; другие в неонатальном или младенческом возрасте поступают в клинику с дыхательной или сердечной недостаточностью. Сдавление бронхов приводит к гипервентиляции и рецидивирующему неонатальному пневмотораксу с дальнейшим ухудшением состояния. Большинство больных требуют хирургического вмешательства в неонатальном периоде или младенческом возрасте.
Хирургическое лечение
Исторически хирургическое лечение было направлено на устранение аневризматического расширения легочных артерий, однако на протяжении многих лет не был выработан однозначный подход к лечению этой патологии. Некоторые хирурги применяли суживание или перевязку легочной артерии для уменьшения их пульсации с наложением анастомоза по Blalock-Taussig или без него. Применялись также ушивание аневризм, аневризморрафия с подвешиванием легочной артерии к ретростернальной фасции. Описан метод транслокации легочной артерии из заднего средостения в переднее с помощью протеза. Некоторые авторы как дополнение к полной коррекции порока предлагают использовать маневр Lecompte — перемещение легочной артерии кпереди от аорты и далеко от трахеобронхиального дерева.
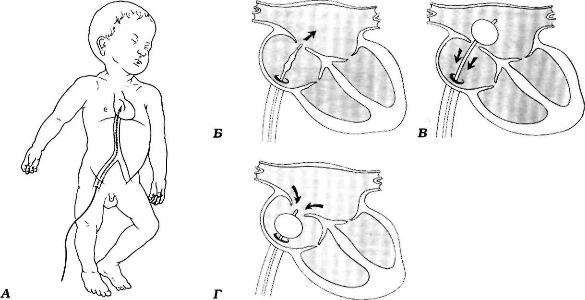
Ни один из этих паллиативных методов не был достаточно эффективным. Хирургическая летальность была достаточно высокой, однако последние сообщения об использовании аортальных гомографтов демонстрируют улучшение результатов. В течение последнего десятилетия по мере накопления опыта коррекции различных пороков в неонатальном возрасте большинство хирургов предпочитают одноэтапное лечение. Оно предусматривает имплантацию гомоклапана, кондуита или моностворки в легочную позицию, закрытие заплатой ДМЖП и гофрирование одной или обеих легочных артерий. Имплантация клапана даёт лучший гемодинамический результат, о чем свидетельствует улучшение физической работоспособности и функционального состояния правого желудочка. Имеется много сообщений о полной коррекции порока с имплантацией механического клапана и хорошим клиническим результатом.
Иногда может потребоваться стентирование крупных бронхов при тяжелом сужении. При выраженном растяжении легкого может возникнуть необходимость удаления пораженного сегмента или лобэктомии. Corno и соавторы устраняли компрессию бронха после коррекции порока путем пересечения аневризматически расширенной легочной артерии и её удлинения с помощью кондуита.
Главным фактором, определяющим летальность, является выраженность легочных проявлений синдрома. Дооперационная ангиографическая оценка аномалий ветвления сегментарных легочных артерий очень важна для прогнозирования исхода. Сосудистые изменения могут оказаться настолько выраженными, что они не совместимы с жизнью как после операции, так и без неё.
У пациентов без симптоматики или с невыраженными проявлениями дыхательной недостаточности операцию обычно откладывают до старшего возраста, когда хирургическая летальность значительно ниже.
Результаты хирургического лечения
Педиатрический консорциум, занимающийся вопросами лечения заболеваний сердца, представил данные об исходах лечения 41 пациента с тетрадой Фалло и отсутствующим клапаном легочной артерии. Двадцать пациентов умерли. Большинство из них были в неонатальном или младенческом возрасте, с массой тела менее 5 кг. В другом сообщении из 28 оперированных в возрасте от 1 дня до 16 лет 6 умерли в раннем послеоперационном периоде и 1 — в отдаленные сроки. Выживаемость к 1 году составила 77% и к 10 годам — 72%. Трое пациентов подвергнуты повторным операциям из-за сохраняющейся дыхательной недостаточности, которая была устранена после повторной аневризморрафии и имплантации клапанного кондуита. Фактором риска была необходимость в дооперационной интубации.
Следует отметить большой процент неизбежных повторных операций после имплантации механических клапанов, а также по поводу стенозов в случаях агрессивной резекции или суживания легочных артерий.
Редкие осложнения
Описаны случаи сдавления передней нисходящей коронарной артерии аневризмой легочного ствола, массивное легочное кровотечение, обструкция легочных вен расширенной ветвью легочной артерии.
После операции параметры вентиляции и газообмена в покое и при максимальной нагрузке ниже нормы. Дыхательный резерв несколько ниже у пациентов с отсутствующим легочным клапаном, чем у больных с обычной формой тетрады Фалло. Это связано с более высоким соотношением мертвого пространства и объёма вентиляции при максимальной нагрузке. Таким больным необходима большая минутная вентиляция для достижения адекватной альвеолярной вентиляции.
У пациентов, перенесших коррекцию порока в младенческом возрасте, часто имеются реактивные дыхательные пути с хронической обструкцией, и поэтому они нуждаются в длительной бронхолитической терапии. У некоторых больных после резекции или пликации аневризмы рост легочных артерий отстает, поэтому возникает необходимость баллонной ангиопластики и эндоваскулярного стентирования. В послеоперационном периоде большинство тяжелых новорожденных нуждаются в экстракорпоральной мембранной оксигенации.
Одной из типичных дополнительных аномалий является недоразвитие миокарда правого желудочка. Трехстворчатый клапан в этих условиях может быть стенозированным или неперфорированным, как при АТК. При этом пороке боталлов проток персистирует, в отличие от тетрады Фалло с отсутствующим клапаном легочной артерии.
Характерным признаком является рыхлость миокарда, межжелудочковая перегородка может быть непропорционально утолщена, напоминая асимметрическую гипертрофию.
При сочетании отсутствующего клапана легочной артерии с АТК прогноз плохой. Больные подлежат только паллиативному лечению в виде межсосудистого анастомоза или операции Fontan.








- Аллергия
- Ангиология
- Болезни глаз
- Венерология
- Гастроэнтерология
- Гинекология
- Дерматология
- Здоровое питание
- Инфекционные болезни
- Кардиология
- Косметология
- Лекарства
- Лекарственные растения
- ЛОР-заболевания
- Мужское здоровье
- Неврология
- Неотложная помощь
- Новости
- Онкология
- Ортопедия
- Паразитология
- Педиатрия
- Пульмонология
- Расшифровка анализов
- Симптомы
- Системные заболевания
- Стоматология
- Травматология
- Урология
- Хирургия
- Эндокринология
- Нужно знать
- Еда
- Профессиональные заболевания
Комментарии